Физико-химические основы коагулирования примесей воды
СОДЕРЖАНИЕ: Седиментация под действием сил тяжести - широко применяемый прием снижения содержания взвеси в воде. Технологический процесс коагуляции примесей. Гидролиз железного купороса в воде. Защита гидрофобных коллоидов, с точки зрения технологии очистки воды.Физико-химические основы коагулирования примесей воды
Седиментация под действием сил тяжести
Одним из наиболее широко применяемых на практике приемов снижения содержания взвеси в воде является седиментация под действием сил тяжести. Однако, примеси, обусловливающие мутность и цветность природных вод, отличаются малыми размерами, вследствие чего их осаждение происходит крайне медленно, так как силы диффузии превалируют над силами тяжести. Кроме того, наличие примесей коллоидного характера еще более осложняет процесс седиментации. Для ускорения процессов осаждения, фильтрования, флотации и повышения их эффективности прибегают к коагулированию примесей воды.
Коагуляцией примесей воды называется процесс укрупнения коллоидных и взвешенных частичек дисперсной системы, происходящей в результате их взаимодействия и объединения в агрегаты. Завершается этот процесс отделением агрегатов слипшихся частичек от жидкой фазы.
Диспергированные, коллоидные и взвешенные частички примесей природных вод в большинстве случаев имеют одинаковые заряды, что обусловливает возникновение межмолекулярных сил отталкивания и агрегативную устойчивость. Поскольку в технологии очистки воды предусматривается частичное или полное удаление примесей, агрегативную устойчивость частичек стремятся нарушить, а заряд их устранить или снизить до очень малых значений. Этого достигают добавлением к воде сульфатов алюминия, железа (II) и железа(III), хлорида алюминия, хлорида железа(III), алюмината натрия, оксихлорида алюминия и других веществ, которые, являясь коагулянтами, либо нарушают агрегативную устойчивость системы, либо образуют вследствие гидролиза коллоиды, сорбирующие примеси из воды.
Коллоидные примеси, находящиеся в природной воде, позволяют рассматривать ее как гетерофазную систему, в которой вода является дисперсионной средой, а масса распределенных в воде коллоидных частичек — дисперсной фазой. Эти частички представляют собой очень мелкие агрегаты кристаллического или аморфного строения. Благодаря огромной удельной поверхности коллоидных частичек они обладают весьма значительной поверхностной энергией, а следовательно, и высокой адсорбционной емкостью. Это обстоятельство имеет большое значение, поскольку основной процесс обработки воды — коагулирование — связан с адсорбцией на коллоидных частичках примесей, содержащихся в воде.
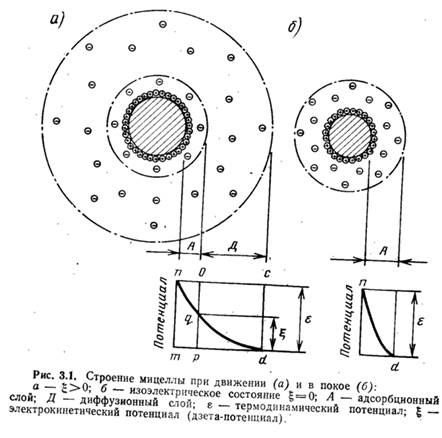
Возникновению коллоидных частичек предшествует образование твердой фазы (ядра), адсорбирующей из растворов потенциалобразующие ионы. Сильнее адсорбируются ионы, которые больше понижают свободную энергию поверхности твердой фазы. В результате поглощения ионов поверхность ядра приобретает заряд. Находящиеся в растворе разноименно заряженные ионы (противоионы) собираются у поверхности ядра вследствие электрического притяжения между разноименными электрическими зарядами, образуя коллоидную частичку. Если бы в растворе не было теплового движения, приводящего к перемещению ионов, противоионы образовали бы мономолекулярный слой (рис. 3.1,б), охватывающий коллоидную частичку на расстоянии ионного радиуса. Термодинамический потенциал у такого двойного слоя является потенциалом между твердой фазой и жидкостью. В действительности упорядоченное строение оболочки нарушается в результате теплового движения ионов в растворе, а слой окружающих частичку противоионов приобретает диффузный характер (см. рис. 3.1, а).
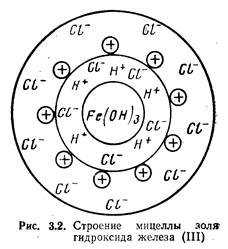
Коллоидная частичка вместе с окружающим ее диффузным слоем называется мицеллой. На рис. 3.2 представлена схема строения мицеллы золя Fe(OH)3, полученного вследствие гидролиза FeCl3. Золь — система, состоящая из коллоидных частичек, распределенных в жидкой среде. Если такой средой является вода, система называется гидрозолем. Как видно из рис. 3.2, мицелла Fe(OH)3 состоит из ядра, образованного молекулами Fe(OH)3, адсорбционносвязанных с ним потенциалобразующих водородных ионов (Н+) и некоторого количества ионов хлора С1 меньшего, чем количество ионов Н+, в результате чего коллоидная частичка имеет положительный заряд. Ионы Н+ и входящие в состав частички противоионы С1- образуют двойной электрический слой. Отдельные ионы хлора (С1-) образуют диффузный слой и вместе с коллоидной частичкой составляют мицеллу золя Fe(OH)3
В зависимости от условий образования золя потенциаловра- зующие ионы и противоионы могут меняться местами.
Золь А1(ОН)3, образующийся при гидролизе солей алюминия, заряжен положительно при низких значениях рН воды и отрицательно — при высоких. Поверхность коллоидной частички золя приобретает заряд в результате нескольких возможных процессов: в щелочной среде кристаллическая решетка частички достраивается гидроксильными ионами, находящимися в воде, приобретая отрицательный заряд; в нейтральной и кислых средах — положительный заряд, который возникает за счет адсорбции решеткой ионов А13+.
При гидролизе растворов Al2(S04)3 потенциалобразующими ионами и противоионами будут соответственно ионы А13+ и S042-.
Известно, что коллоидные частички находятся в постоянном движении. При этом часть окружающего частичку раствора увлекается и движется вместе с ней в виде тонкой пленки. На рис. 3.1, а линия 00 представляет поверхность коллоидной частички, непосредственно возле которой расположены положительные ионы двойного слоя, а далее ионы диффузного слоя, ограниченные на рисунке линией CD, являющейся границей электронейтрального комплекса мицеллы. При движении коллоидная частичка в электрическом поле увлекает часть раствора, ограниченную на рисунке линией АВ, а часть ионов диффузного слоя, расположенных между линиями АВ и CD, отрывается от частички. При этом она становится отрицательно заряженной, а окружающий ее раствор приобретает положительный заряд. Скачок потенциала, возникающий при этом между частью жидкости, увлекаемой коллоидной частичкой, и остальным раствором, называется электрокинетическим, или потенциалом.
g-потенциал изменяется при прибавлении к коллоидным системам электролитов. Для отрицательно заряженных частичек потенциал зависит от величины заряда катионов электролита, а для положительно заряженных частичек — от величины заряда анионов.
С прибавлением электролитов концентрация ионов в диффузном слое увеличивается, и для компенсации зарядов на поверхности частичек требуется меньший объем диффузного слоя (т. е. происходит как бы его сжатие). Сжатие может дойти до такой степени, что диффузный слой не будет выходить за пределы линии, ограничивающей поверхность скольжения коллоидной частички при ее движении (см. рис. 3.1,б линия АВ). Когда диффузный слой сожмется до размера, ограниченного линией АВ, g-потенциал станет равным нулю. В этот момент коллоидные частички будут находиться в изоэлектрическом состоянии (изоэлектрическим состоянием называется состояние золя, при котором коллоидные частички не имеют электрического заряда). В результате устранятся причины, препятствовавшие их сближению, коллоидные частички, соединяясь, образуют сравнительно крупные агрегаты, которые начинают осаждаться.
При добавлении электролита к коагулируемому коллоиду можно заметить, что коагуляция начинается не в изоэлектрической точке, а при значении потенциала 0,03 В (значение потенциала для большинства коллоидов обычно составляет 0,07 В). Это значение потенциала является мерой устойчивости коллоидных систем и называется критическим; с его уменьшением устойчивость коллоидной системы снижается.
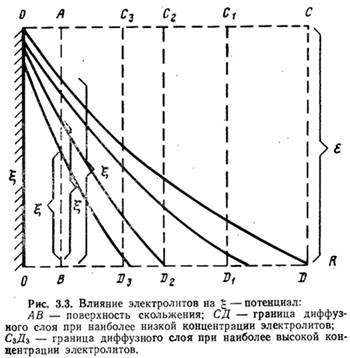
Изменение значения g-потенциала при сжатии диффузного слоя показано на рис. 3.3 (по оси абсцисс отложены расстояния от поверхности частички, по оси ординат — значения потенциалов). Степень влияния ионов зависит от их концентрации, валентности и размеров: чем выше концентрация ионов и их валентность, тем больше сжатие диффузного слоя, а следовательно, тем сильнее эти ионы снижают устойчивость коллоидных частичек.
Так происходит коагуляция гидрофобных золей, обусловленная адсорбцией ионов и созданием двойного электрического слоя на поверхности коллоидных частичек. Их устойчивость в растворе определяется гидратацией ионов и влиянием зарядов поверхности на ориентированную адсорбцию дипольных молекул воды. Эти гидратные слои полностью зависят от ионных взаимодействий и при электролиткой коагуляции не препятствуют слипанию частичек.
Совершенно иной характер устойчивости имеют гидрофильные золи, природа поверхности частичек которых обусловливает образование молекулярных сольватных слоев при участии вандерваальсовских, водородных и комплексных связей, вне зависимости от действия растворов электролитов небольших концентраций. Высокоочищенные золи H2SiO3 и А1(ОН)3 могут сохраняться в растворе даже при снижении g-потенциала почти до нуля.
Значение гидратных слоев объяснено тем, что для сближения коллоидных частичек необходимо затратить работу на преодоление сопротивления так называемого «расклинивающего давления», обусловленного силами молекулярного сцепления воды с поверхностью частичек. На расстоянии 1 нм и меньше силы взаимного притяжения частичек преобладают над силами сцепления в гидратном слое. При больших расстояниях гидратные слои являются термодинамически устойчивым стабилизирующим фактором.
Аналогично влияют поверхностно-активные вещества. Изменение гидрофильности поверхности частичек в данном случае зависит от ориентации молекул поверхностно-активных веществ в адсорбционном слое. Увеличение гидрофильности и возрастание устойчивости коллоидных частичек в водных системах наблюдается, если полярные группы адсорбированных соединений обращены в сторону дисперсионной среды. Ориентация приобретает особое значение при образовании молекулами поверхностно-активных веществ в адсорбционных слоях двухмерных гелеобразных структур, обладающих повышенными структурно-механическими свойствами. Это явление называется коллоидной защитной, которая заключается в том, что при добавлении гидрофильных веществ к гидрофобным коллоидам они образуют структурно-прочные адсорбционные слои на поверхности частичек и повышают устойчивость последних по отношению к электролитам-коагулянтам.
Добавление к золю небольших количеств высокомолекулярных соединений (ВМС), которые не обеспечивают полного покрытия поверхности частичек золя, вызывает явление, противоположное коллоидной защите, — сенсибилизацию, т. е. повышение чувствительности золя к действию электролитов. Сенсибилизирующее действие ВМС проявляется независимо от знака заряда поверхности частичек золей. Термодинамическая устойчивость таких растворов определяется тем, что связь молекул полимерного соединения с водой сильнее их взаимной связи в твердой фазе и тем, что они равномерно распределены во всем объеме растворителя.
При очистке воды коагуляция коллоидов протекает под влиянием сложной смеси электролитов, находящихся в воде, и под влиянием ионов, вносимых в воду вместе с коагулянтом. Так, в случае применения Ai2(S04)3, в воду вносят значительные количества ионов S042-.
Если в качестве коагулянта используется FeCl3, в очищаемой воде повышается содержание ионов С1-. Наличие смеси электролитов усложняет коагуляцию, поскольку при этом эффекты влияния отдельных коагулирующих ионов усиливаются или ослабляются.
Коагуляция коллоидов вызывается не только электролитами, но и взаимодействием противоположно заряженных коллоидов, наступающим при адсорбции одного коллоида поверхностью другого. Этот процесс играет некоторую роль при осветлении воды коагулированием. Необходимым условием взаимной коагуляции является равенство противоположных зарядов частичек золей. При несоблюдении этого условия коагуляция протекает либо неполно, либо вовсе не наступает, независимо от количества прибавленного коагулирующего коллоида. Отсюда следует, что взаимная коагуляция коллоидов может протекать лишь в узкой зоне соотношения их концентраций.
Большинство коллоидов природных вод в отличие от золей коагулянтов заряжено отрицательно. К таким коллоидам относятся распространенные в природных водах кремниевая кислота, мельчайшие глинистые и почвенные частички, а также гумусовые вещества. Глинистые и почвенные взвеси состоят в основном из гидроалюмосиликатов.
Вода, входящая в состав глины, по современным представлениям не является кристаллогидратной. Молекула глины представляет собой молекулу алюмосиликатной кислоты, в которой кислород воды входит в состав аниона, а водород является катионом; простейшая формула гидроалюмосиликата в этом случае выглядит так.
Анионы алюмосиликата образуют основу отрицательно заряженной глинистой частички, окруженной сферой положительных компенсирующих ионов водорода. Величина заряда глинистой частички и его знак зависят от рН воды, в которой суспензирована глина, поскольку при повышении концентрации, ионов водорода уменьшается диссоциация алюмосиликатной кислоты, а благодаря этому и количество свободных ионов, обусловливающих устойчивость коллоидных частичек. Опыт подтверждает, что глинистые взвеси значительно скорее отстаиваются и коагулируют при подкислении воды.
Гумусовые вещества, также имеющие кислотный характера в природных водах встречаются в виде стойких высокодисперсных отрицательно заряженных золей. Основу частичек составляют анионы, гуминовых кислот; ионы водорода или металлов, составляют внешнюю оболочку двойного электрического слоя.
Большинство веществ, обусловливающих мутность и цветность природных вод, являются гидрофобными, или слабогидрофильными коллоидами. Гидрофобны или слабогидрофильны и коллоиды, образующиеся при очистке воды в результате гидролиза вводимых в воду коагулянтов. Поэтому полное представление о поведении коллоидов в процессе очистки природных вод можно получить лишь при учете факторов устойчивости гидрофобных и гидрофильных коллоидов.
На наличие гидрофильных свойств у коллоидов природных вод указывают три фактора: малая чувствительность к содержанию электролитов; способность удерживать значительное количество воды их скоагулированными осадками; для некоторых из них, в особенности для продуктов разложения белковых веществ и гуминов, характерно проявление защитного действия по отношению к глинистым и почвенным суспензиям, коллоидной кремниевой кислоте, а также к золям гидроксидов алюминия и железа (III), образующихся при внесении в воду коагулянтов.
Защита гидрофобных коллоидов, с точки зрения технологии очистки воды, явление нежелательное, поскольку она вызывает образование стойких связей и замедляет осаждение коагулянта.
Коллоидные частички гидрофильных органических коллоидов представляют собой агрегаты молекул, свернутых в рыхлый клубок, промежутки в котором заполнены водой. Полярными группами в них (например, в гуминовых веществах) являются фенольные и карбоксильные. Устойчивость гидрофильных коллоидов объясняется развитой гидратной оболочкой. Например, у полярных групп ОН-, С032_, НС03-, образующих коллоидные гидрофильные частички, молекулы воды, представляющие собой диполи, ориентируются и притягиваются в результате электростатического взаимодействия. По мере удаления от поверхности полярных молекул ориентация молекул воды ослабевает. Устойчивость гидрофильных систем снижается при химической обработке, в результате которой полярные группы НСО3- окисляются до карбонильных — гидрофобных (количество полярных групп в молекуле уменьшается). Для этой цели применяют предварительное хлорирование воды перед введением коагулянта.
Используемые в технологии очистки воды коагулянты чаще всего являются солями слабых оснований а сильных кислот (A12(S04)3, FeS04, FeCl3 и др.). При растворении они гидролизуют. Взаимодействуя с гидроксильными ионами, содержащимися в воде, в результате электролитической диссоциации последней эти соли образуют малорастворимые основания. В воде накапливаются ионы водорода, и раствор приобретает кислую реакцию.
Полнота гидролиза имеет большое значение как для самой коагуляции, так и для качества очищаемой воды, поскольку наличие ионов алюминия в очищенной питьевой воде недопустимо.
Скорость гидролиза можно описать уравнением:
![]()
где v— скорость гидролиза; Кг — константа гидролиза; [Мел+] — концентрация катионов в растворе коагулянта; [Н20] — концентрация воды в растворе.
Из уравнения следует, что скорость гидролиза пропорциональна концентрации катионов коагулянта. Поскольку применяющиеся концентрации растворов коагулянта незначительны, можно считать, что скорость гидролиза коагулянта прямо пропорциональна его концентрации (или его дозе), введенной в воду. Согласно правилу Вант—Гоффа с повышением температуры на каждые 10 градусов скорость гидролиза, как и большинства химических реакций, возрастает примерно в 2—4 раза.
Необходимым условием для более полного протекания гидролиза является удаление из сферы реакции образующихся Fe(OH)3 или А1(ОН)3, а также связывание ионов Н+ в малодиссоциированные молекулы. Гидролиз усиливается с разбавлением коагулянта.
Более полному гидролизу подвержены коагулянты, образующие гидроксиды с меньшей константой диссоциации (величиной, характеризующей способность электролитов диссоциировать на ионы) или меньшим произведением растворимости.
Гидролиз солей железа (III) протекает полнее, чем солей алюминия, и значительно полнее, чем солей железа (II).
Как следует из уравнения, с увеличением степени гидролиза рН раствора должно уменьшаться. Любое повышение рН раствора обеспечивает полный гидролиз введенного в воду коагулянта. Для быстрого и полного гидролиза коагулянтов необходим некоторый щелочной резерв воды, т. е. наличие в ней определенного количества ионов НС03-, ОН-, которые связывают ионы водорода, выделяющиеся при гидролизе:
![]()
Благодаря наличию в воде буферной системы НС03-—Н2СО3 с рН, близким к 7, рН воды при гидролизе коагулянтов в большинстве случаев изменяется незначительно (уменьшается).
По уравнению гидролиза коагулянтов можно вычислить концентрацию ионов НС03-, необходимую для нейтрализации кислоты, образующейся при гидролизе определенной дозы коагулянта. Из суммарной реакции гидролиза Al2(S04)3 в присутствии ионов НС03-
Al2 (S04)3 + ЗСа (НС03)2 + 6Н20 = 2А1 (0Н)3 + 3CaS04 + 6Н2СО
следует, что на каждые 342 м г A12(S04)3 расходуется 6 мг-экв НСОз-.
В тех случаях, когда концентрация ионов ОН-, НС03~, содержащихся в воде, недостаточна для полного гидролиза коагулянта, щелочность воды повышают введением известкового молока или раствора соды. В первом случае эффект подщелачивания объясняется связыванием ионов Н+ ионами ОН- извести, во втором — связыванием ионов Н+ в ионы НС03-.
Зная щелочность обрабатываемой воды и дозу введенного коагулянта, можно вычислить дозу извести или соды, необходимой для гидролиза и обеспечения резервной остаточной щелочности, равной 1 мг-экв/л.
Гидролиз солей алюминия, используемых в качестве коагулянтов, протекает в несколько стадий:
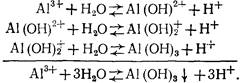
Степень гидролиза повышается с разбавлением раствора, повышением его температуры и рН.
Гидроксид алюминия является типичным амфотерным соединением, т. е. обладает как кислотными, так и основными свойствами:
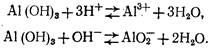
Константа равновесия для первого уравнения
![]()
(А1(ОН)3 содержится в твердой фазе и концентрация его в растворе постоянна), откуда [Al3+]=k[H+]3.
Для второго уравнения константа равновесия:
![]()
Или
![]()
откуда
![]()
седиментация примесь коагуляция гидролиз
где к® — ионное произведение воды (величина постоянная, зависящая только от температуры).
Таким образом, растворимость А1(ОН)3 в кислой среде, прямо пропорциональна концентрации водородных ионов в третьей степени [Н+]3, а в щелочной — обратно пропорциональна [Н+].
Осаждение А1(ОН)3 начинается при рН=3 и становится полным при рН=7. Дальнейшее повышение рН ведет к растворению осадка (пептизации), заметному при рН=9. Рентгенографически установлено, что в гидрокарбонатно-хлоридных и гидрокарбонатно-сульфатных средах частички формируются с образованием гидроаргиллита —А1(ОН)3, способного существовать в этих условиях длительное время.
При коагулировании в кислых и нейтральных средах, содержащих небольшое количество гидрокарбонатов, для нейтрализации ионов Н+, накапливающихся в воде при гидролизе A12(S04)3, можно применять смесь A12(S04)3 и NaA102. В этом случае накапливающиеся ионы Н+ будут нейтрализоваться ионами ОН-, образующимися при гидролизе NaAl02:
![]()
Если в смеси выдержано необходимое соотношение между A12(S04)3 и NaA102, то при гидролизе значение воды практически не изменяется и гидролиз обоих реагентов протекает достаточно полно.
Гидролиз FeCI3 протекает в три стадии:
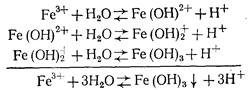
Гидролиз железного купороса в воде выражается уравнением
![]()
Поскольку растворимость Fe(OH)2 велика и он значительно диссоциирован, равновесие гидролиза сильно смещено влево. Образующийся Fe(OH)2 под действием кислорода, растворенного в воде, окисляется в Fe(OH)3:
![]()
Хлопья FeCbнаиболее интенсивно образуются при рН=5... ... 7, причем оптимум находится между значениями рН 6,1 и 6,5. Изоэлектрическая точка Fe(OH)3 соответствует значению рН несколько большему, чем 6,5. Хлопьеобразование Fe(OH)3, в отличие от Аl(ОН)3, протекает в значительно более широком диапазоне значений рН. Рентгенографически установлено, что при гидролизе солей железа (III) в гидрокарбонатно-хлоридных и гидрокарбонатно-сульфатных средах образуется одна и та же модификация гидроксида железа(III) — гетитa-FeO(OH). Характерно, что a-гидроксид железа(III) образуется и в случае гидролиза солей железа(II), например, железного купороса, окисляемого хлором или растворенным в воде кислородом при подщелачивании известью.
Для нормальной коагуляции большую роль играют размеры и структура частичек гидроксидов, причем размеры частичек, выделяющихся при гидролизе коагулянтов, зависят главным образом от степени пересыщения раствора. В процессе водоочистки гидролиз солей-коагулянтов протекает в разбавленных растворах, что создает условия слабого пресыщения и благоприятствует образованию крупных частичек при обеспечении необходимого периода времени на их формирование. Структура выделяющихся из раствора частичек зависит от скорости двух протекающих одновременно процессов: беспорядочного слипания частичек при столкновениях (агрегации) и роста кристаллов (ориентации). При незначительном пересыщении главную роль играет скорость ориентации, которая у гидроксидов понижается с увеличением числа гидроксильных ионов, связанных с атомом металла. Поэтому частички гидроксидов двухвалентных металлов имеют кристаллическую форму, а гидроксиды трехвалентных металлов (алюминия и железа) выделяются главным образом в аморфном состоянии.
Процесс искусственного обесцвечивания воды согласно современным представлениям протекает следующим образом. При добавлении к очищаемой воде раствора коагулянтов в течение первых 30—180 с происходит гидролиз добавленных солей и образуются коллоидные гидроксиды алюминия и железа, имеющие огромные активные поверхности. Коллоидные примеси, содержащиеся в воде, адсорбируются на поверхности частичек Гидроксидов. При адсорбции следует различать два процесса: собственно адсорбцию и фиксацию (закрепление) адсорбированных коллоидов на поверхности. В первом процессе главную роль играют силы межмолекулярного взаимодействия. Адсорбция коллоидных частичек зависит от их дисперсности: она тем больше, чем выше дисперсность и чем меньше устойчивость частичек.
Причины фиксации могут быть различными. Чаще всего необходимость процесса адсорбции гуминов и других коллоидных загрязнений воды на поверхности гидроксидов вызывается образованием особого рода поверхностных соединений — лаков. Большое значение в процессе фиксации адсорбированных коллоидов имеет их коагуляция вследствие разноименности зарядов адсорбированных частичек и поверхности адсорбента. Наличие заряда у адсорбирующихся коллоидных частичек влияет на их адсорбируемость.
Коагуляция частичек Fe(OH)3 и А1(ОН)3, а также связанное с этим выделение их из воды совместно с адсорбированными на их поверхности коллоидными примесями происходит под действием растворенных в воде электролитов. В связи с этим очистка цветных вод с повышенной степенью минерализации протекает обычно лучше, чем мягких, бедных солями вод. Из описанного процесса обесцвечивания следует, что коагуляции подвергаются не коллоидные примеси воды, а образующиеся при гидролизе коагулянтов гидроксиды. Вода очищается не в результате коагуляции, а вследствие адсорбции различных коллоидных и высокомолекулярных примесей на поверхности гидроксидов. Процесс коагуляции гидроксидов фактически приводит к удалению отработанного сорбента из очищенной воды.
Характер осветления природных вод определяется свойствами взвеси: при наличии крупных частичек вода осветляется благодаря их декантации под влиянием силы гравитации, а при наличии высокодисперсных частичек осветление воды определяется их обменной катионной емкостью. Если эта емкость превышает 250 мг-экв/л, вода осветляется без добавления коагулянта в результате сжатия двойного электрического слоя за счет обмена одновалентных ионов на двух- и трехвалентные. Природные воды обычно содержат взвесь со значительно меньшей обменной емкостью. В этом случае эффективное хлопьеобразование наступает лишь при добавлении коагулянта, образующего гидроксид, к хлопьям которого прилипают частички взвеси, или он сам обволакивает взвешенные вещества. Большое значение имеет также ортокинетическая коагуляция вследствие захватывания взвеси сеткой оседающих хлопьев гидроксида. Из сказанного следует, что процесс коагулирования зависит прежде всего от солевого состава воды, главным образом от ее анионного состава, поскольку Fe(OH)3 и А1(ОН)3 заряжены положительно и коагулирующими ионами для них являются анионы. Самыми распространенными анионами большинства природных вод являются НСОз~, С1~ и S042-. Концентрация этих анионов, обеспечивающая максимальную скорость коагуляции Fe(OH)3 и А1(ОН)3, составляет: для SO*2- — 0,001—0,002 н„ для С1- — 0,07 н. и для НС03- — 0,005 н. В природных водах концентрации указанных анионов обычно ниже, следовательно, коагуляция гидроксидов протекает медленнее.
ЛИТЕРАТУРА
1. Алексеев Л.С., Гладков В.А. Улучшение качества мягких вод. М., Стройиздат, 1994 г.
2. Алферова Л.А., Нечаев А.П. Замкнутые системы водного хозяйства промышленных предприятий, комплексов и районов. М., 1984.
3. Аюкаев Р.И., Мельцер В.3. Производство и применение фильтрующих
4. материалов для очистки воды. Л., 1985.
5. Вейцер Ю.М., Мииц Д.М. Высокомолекулярные флокулянты в процессах очистки воды. М., 1984.
6. Егоров А.И. Гидравлика напорных трубчатых систем в водопроводных очистных сооружениях. М., 1984.
7. Журба М.Г. Очистки воды на зернистых фильтрах. Львов, 1980.