Ионно-парная хроматография
СОДЕРЖАНИЕ: Сущность и содержание ионно-парной хроматографии, ее использование в жидкостной хроматографии и экстракции для извлечения лекарств и их метаболитов из биологических жидкостей в органическую фазу. Варианты ионно-парной хроматографии, отличительные черты.Ион-парная хроматография
Ион-парная хроматография давно находила применение в жидкостной хроматографии и экстракции для извлечения лекарств и их метаболитов из биологических жидкостей в органическую фазу. Как самостоятельный раздел ВЭЖХ ион-парная хроматография, называвшаяся также экстракционной, парно-ионной, хроматографией с использованием ПАВ, хроматографией с жидким ионообменником, стала развиваться с середины 70-х годов. Метод занимает промежуточное положение между ионообменной хроматографией и адсорбционной, распределительной или обращенно-фазной. Недостатки ионообменных материалов, а именно невоспроизводимость от партии к партии, меньшая активность и стабильность по сравнению с другими сорбентами и небольшой выбор наполнительного материала, исключающий изменение селективности за счет сорбента, привел к некоторому ограничению применения ионообменной хроматографии. В ион-парной хроматографии большинство этих недостатков можно преодолеть. Метод ион-парной хроматографии характеризуется универсальностью и обладает преимуществом по сравнению с классической ионообменной хроматографией, в котором активные центры фиксированы. Вследствие более быстрой массопередачи в ион-парной системе хроматографическое разделение более эффективно, чем на ионообменнике с фиксированными и активными зонами.
Ион-парную хроматографию используют для разделения образцов, содержащих как ионные, так и неионные соединения. Ее применяют в тех случаях, когда трудно или невозможно получить приемлемое разделение образца методом ионообменной хроматографии адсорбционной или обращенно-фазной. В некоторых случаях ионные соединения можно разделить на обращенной фазе, придавая им свойства неионных соединений (подавление ионов) с помощью буферного раствора с соответствующим рН, при котором равновесие смещается в сторону образования неионизированной формы. Полярные вещества, обладающие липофильными свойствами, делятся при этом на обращенной фазе как неполярные. Однако большинство наполнительных материалов колонок надежно работает только при рН=1,5–7,5. Исключение составляет партисил 5 ОДС, работающий при рН=1–8,5. В этом диапазоне рН сильные кислоты и основания ионизированы.
Попытки разделения сильных кислот и оснований методом подавления ионов оказываются неудачными из-за плохого удерживания веществ и асимметрии пиков. Соединения, остающиеся ионизированными в интервале рН=2–8, удовлетворительно разделяются методом ион-парной хроматографии, когда в подвижную фазу добавляют противоион, заряд которого противоположен заряду молекулы, и создается ион-парный комплекс, обладающий свойствами неполярного вещества. Если к ионному соединению, растворимому только в воде, добавить противоион, то образуется ионная пара, которая, обладая свойством растворяться в органической фазе, распределится между водным и органическим слоем. Возможна также адсорбция липофильной части противоиона в углеводородной фазе наполнительного материала. Очевидно, что катионы будут хорошо экстрагироваться анионами, и наоборот.
Таким образом, ионизированные молекулы находятся в равновесии и образуют ионную пару: растворенное вещество–противоион, причем все равновесия имеют концентрационные зависимости. В упрощенном виде распределительное равновесие может быть представлено в виде
В+ вод + Р – орг = (В+ Р- ) орг,
где В+ – протонированная форма основания, которое нужно экстрагировать; Р- – анион кислоты, который применяют для образования ионной пары.
Ионная пара В+Р – будет растворяться в полярной органической фазе, например в смеси спирта с хлороформом, а ионные формы будут растворяться в воде. Для определения ароматических сульфокислот применяют в качестве противоиона тетра-бутиламмоний, а для анализа хинина–сульфокислоты камфоры. В качестве противоиона обычно используют четвертичные или третичные амины, соли сульфокислот. Наиболее часто применяют тетраметил, тетрабутил, пальметилтриметиламмоний для анализа кислот, сульфированных красителей и третичные амины типа триоктиламина для анализа сульфонатов. Противоионами для анализа оснований являются соли алкил- и арилсульфокислот, перхлораты, пикраты.
Существует четыре варианта ионно-парной хроматографии:
1) адсорбционная хроматография, когда ионные пары вымываются элюентом с силикагеля;
2) нормально-фазная распределительная хроматография, когда вода, нанесенная на пористую подложку, является неподвижной фазой, органический растворитель–элюентом;
3) обращенно-фазная распределительная хроматография с органическим растворителем в качестве неподвижной фазы и водой в качестве элюента;
4) обращенно-фазная хроматография, когда гидрофобный ион, образующий ионную пару, адсорбируется углеводородной частью неподвижной фазы. Иногда добавляют ПАВ, например цетилтриметиламмонийбромид (цетримид).
Ион-парную хроматографию применяют и для разделения амфотерных веществ. Когда ион-парную хроматографию применяют в нормально-фазном варианте в качестве противоионов, иногда используют ионы, способные к абсорбции света или к флуоресценции, для улучшения идентификации некоторых не поглощающих свет соединений. В этом варианте ион-парной хроматографии селективность системы изменяется за счет изменения полярности органической фазы. В табл. 3.4 приведены примеры использования ион-парной хроматографии при работе в режиме нормально-фазной хроматографии.
Однако наиболее часто применяют ион-парную хроматографию на обращенной фазе, при которой в качестве подвижной фазы используют водный буферный раствор и органический растворитель, смешивающийся с водой, обычно метанол или ацетонитрил. В подвижную фазу добавляют противоион, заряд которого противоположен заряду молекулы, а в качестве сорбента используют силикагель с химически привитой фазой, обычно С8 или C18. Иногда разделение осуществляют с применением несмешиваемой с водой механически удерживаемой фазы, например, бутанола. При разделении на обращенной фазе более стабильной, чем механически удерживаемая фаза, водные образцы могут непосредственно вводиться в колонку, что особенно важно для анализа биологических образцов. При этом нет необходимости в предварительной очистке, так как гидрофильные компоненты мгновенно вызываются из колонки. Градиентное элюирование проводят, изменяя концентрацию противоиона в подвижной фазе или меняя полярность растворителя. При изменении концентрации противоиона, который остается в неподвижной фазе, изменяется сила растворителя, а при изменении рН подвижной фазы изменяется селективность разделения.
От обычной обращенно-фазной хроматографии легко перейти к ион-парной на обращенной фазе, и наоборот.
Ион-парное разделение на обращенной фазе (табл. 3.5) может быть проведено несколькими методами:
1) на привитой к матрице неподвижной фазе, состоящей из углеводородов;
2) то же самое, но в качестве противоиона используют ПАВ;
3) на неподвижной фазе, состоящей из механически удерживаемой органической жидкости;
4) на неподвижной фазе, содержащей жидкий ионообменник.
Важным условием проведения ион-парной хроматографии является стабильность системы. Это означает в случае механически удерживаемой жидкости несмешиваемость водной и органической фаз, что достигается четким термо-статированием и предварительным насыщением подвижной фазы неподвижной. При работе с нормальной фазой при введении противоиона в неподвижную фазу необходимо предотвратить его унос неподвижной фазой за счет образования ионных пар, покидающих болонку. Противоион в этом случае добавляют в образец до введения его в хроматограф или в подвижную фазу. Поскольку в ион-парной хроматографии работают с полярными веществами, склонными к образованию хвостов, следует помнить, что в этом случае желательно применить другую подвижную или неподвижную фазу, другой противоион. Необходимо, чтобы в ион-парной хроматографии при изменении концен-трации не изменялось значение k образца, что может повлечь образование хвостов. Водная фаза должна иметь постоянную концентрацию лротивоиона и рН. Обычно используют цитратный или фосфатовый буферный раствор. Иногда противоион сам является буфером. В случае разделения при низких рН растворы сильных кислот обеспечивают достаточное буферное действие.
Интересно проследить роль противоиона в ион-парной обращенно-фазной хроматографии. Можно написать следующие уравнения для образца, имеющего анион и
k=(Vs/Vm) E(c+ ), k=(Vs/Vm) E (c-),
где Е–константа экстракции конкретной ион-парной системы; (с+) и (с-) – концентрации анионного и катионного противоиона.
При прочих неизменных условиях Е постоянна и, следовательно, повышение концентрации противоиона в подвижной фазе приводит к увеличению k при разделении на обращенной фазе. В нормально-фазной ион-парной хроматографии k также меняется за счет изменения концентрации противоиона в подвижной фазе. Значение k может регулироваться типом противоиона, например, замена гептансульфокислоты пентансуль-фокислотой может изменить k в 2–5 раз. Этот эффект ярко выражен при низких концентрациях противоиона. Крупные молекулы противоиона дают большие величины k при ион-парном разделении на обычной фазе. Так, переход от тетра-этиламмония к тетрапентиламмонию позволил изменить k на несколько порядков.
Способность различных анионов экстрагировать ион тетра-бутлламмония из воды в хлороформ является мерой эффективности этих противоионов (табл. 3.6).
В тех случаях, когда вещество полностью ионизировано, изменение силы растворителя за счет изменения концентрации противоиона не влияет на селективность, и только когда вещество частично ионизировано или не ионизировано, селективность меняется при изменении концентрации противоиона.
В ион-парном разделении на обращенной фазе сила растворителя меняется за счет изменения полярности подвижной фазы. Увеличивая в смесях воды с метанолом или ацетонитри-лом содержание воды, мы увеличиваем силу растворителя и снижаем значение k для образца. В ион-парной хроматографии в качестве подвижных фаз применяют бутанол, пентанол, метиленхлорид и гексан. При этом более полярные растворители являются более сильными и дают самые низкие значения k. Сила растворителя в ион-парной хроматографии зависит от его способности стабилизировать или растворять ионы и ионные пары, в отличие от фактора полярности растворителя Р, связанного с его способностью растворять полярные неионные вещества. Сила растворителя в ион-парной хроматографии зависит от параметра Р и от его диэлектрической проницаемости е. Показателем относительной силы растворителя служит функция Р+0,25 (табл. 3.7).
Повышение ионной силы водной фазы приводит к уменьшению числа образующихся ионных пар из-за конкуренции буферных ионов с противоионом за образование ионной пары. Поэтому повышение ионной силы в ион-парной хроматографии приводит к снижению k при разделении на обращенной фазе и к повышению k при разделении на нормальной фазе. Влияние буферных ионов возрастает в последовательности: NO2-Br-.Cl-SO42 – Селективность растворителя в ион-парной хроматографии изменяется по тем же правилам, как и в случае распределительной жидкостной хроматографии.
Оптимальными при ион-парном разделении на обращенной фазе являются средние значения рН. При снижении рН подвижной фазы анионы Х – начинают превращаться в неионизированные кислоты и число ионных пар образца в неподвижной фазе уменьшается, а следовательно, снижается и значение k. Изменение рН оказывается мощным средством изменения селективности разделения. При высоких значениях рН значение k’ также падает, что аналогично уменьшению обменной емкости, так как ионы ОН – подвижной фазы начинают связывать противоионы и конкурировать с анионом образца в образовании ионных пар. Слабые кислоты или основания обычно не используют в качестве противоионов для ион-парной хроматографии.
При ион-парном разделении на нормальной фазе зависимость k от рН обратна. Компоненты образца более сильно удерживаются при низких и при высоких значениях рН при условии, что неионизированные ионы образца не удерживаются водной фазой. Объем вводимого в ион-парной хроматографии вещества обычно не должен быть очень большим, чтобы не было размывания зон. Иногда ограничивающим фактором является
концентрация противоиона в подвижной фазе; повышая его концентрацию, можно увеличить максимальную концентрацию вводимого вещества. При повышении концентрации противоиона и соответственном изменении значений k образца возможно одновременное добавление избытка нейтральной соли в водную фазу, что стабилизирует значение k. Получаем закономерность, аналогичную закономерности влияния буферного раствора. Максимальное количество вводимого образца может быть повышено при добавлении образца в виде ионных пар. При этом до введения в хроматограф противоион смешивают с образцом, а рН доводят до нужного значения.
Влияние температуры имеет в ион-парной хроматографии большое значение. При использовании механически удерживаемых неподвижных фаз колонка должна быть термостатирована. В ион-парной хроматографии применяют обычно фазы с повышенной вязкостью, а повышение температуры снижает ее. Зависимость селективности от температуры также наиболее выражена в ион-парной хроматографии.
Применяя противоионы, поглощающие в УФ-области, можно получать при ион-парном разделении легко обнаруживаемые спектрофотометром ионные комплексы. Требуется, однако, чтобы противоионы не растворялись в органической фазе во избежание высокого поглощения выходящего из колонки раствора. Таким образом, используя ион пикрата или 2-нафтилсульфоната, можно обнаружить амины.
Одним из затруднений, наиболее часто встречающихся в ион-парной хроматографии, является нестабильность колонок, особенно в обращенно-фазном режиме. В колонках с обычной фазой наблюдается постепенный унос противоиона из неподвижной фазы, однако этого можно избежать, получая ионные пары до введения образца в хроматограф. Большим недостатком ион-парной хроматографии является образование хвостов. Причиной этого является либо диссоциация ионных пар, которая уменьшается при повышении концентрации противоиона, либо неправильная концентрация буферного раствора. Иногда удается уменьшить затягивание зон и увеличить эффективность разделения, перейдя от обычной ион-парной хроматографии к хроматографии с использованием поверхностно-активных веществ.
Такой способ разделения, по-видимому, пригоден для анализа очень полярных молекул, например сульфированных красителей. Длина углеродной цепи неподвижной фазы также варьируется в ион-парной хроматографии.
Воспроизводимость колонок в ион-парной хроматографии удовлетворительная в отличие от таковой в ионообменной хроматографии.
Ион-парную хроматографию обычно применяют для анализа физиологических и биологических жидкостей, полярных соединений и веществ с несколькими ионизируемыми группами, в том числе промежуточных продуктов красителей. Расфасованные реагенты для ион-парной хроматографии, состоящие из буфера и противоиона, которые можно непосредственно добавлять в подвижную фазу, выпускает фирма «Уотерс». К ним относится реактив А (0,005 М раствор тетрабутиламмонийфосфата, рН=7,5), реактив В-5 (0,005 М раствор пентансульфокислоты, рН=3,5) и реактив В-7 (0,005 М раствор гептансульфокислоты, рН=3,5).
При отсутствии четких литературных аналогий начинают разделение методом ион-парной хроматографии на обращенной фазе C18 с размером частиц 5–10 мкм. Наполнителем в ион-арной хроматографии с добавкой органической неподвижной азы является материал, используемый для обращенной фазы, при работе с нормальной фазой применяют обычный силикагель 5–10 мкм, как и в случае адсорбционной хроматографии. возможно применение нейтральных полистирол-дивинильных смол или смол ХАД. Колонки с C18 служат дольше в ион-парной хроматографии, чем колонки с неподвижной фазой, имеющей более короткую углеводородную цепь. Последующая после «привязывания» фазы силанизация улучшает свойства материала и увеличивает срок его службы (партисил 5 ОДС).
Для увеличения стабильности колонки рН следует уменьшать по мере увеличения концентрации противоиона.
Для этой же цели предложено использовать триэтиламин в качестве основания, так как этот реактив доступен, растворим, удобен в работе и обладает малой химической актив-ностью. Предполагается, что сильные основания, так же как четвертичные гидроксиды, разрушают силикагелевую подложку. Подвижную фазу для ион-парной хроматографии желательно фильтровать через фильтр из стекловолокна, а после окончания работы колонку следует промывать пятикратным объемом элюента метанол – вода (50: 50).
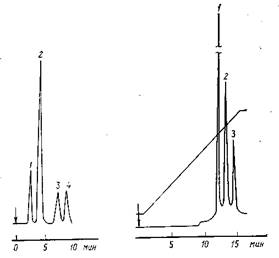
Рис. 1. Хроматограмма витаминов, полученная на колонке размером 300 Х 4 мм с µ-бондапаком C18 , подвижная фаза – метанол – вода (70: 30) с 0,1% В7 и В5 (1:1), расход 1 мл/мин, детектор–УФ (254 нм): 1 – никотинамид; 2 – пиродоксин; 3 – рибофлавин; 4 – тиамин
Рис. 2. Хроматограмма изомеров фталевой кислоты, полученная на колонке размером 300 Х 4 мм с µ-бондапаком C18 , подвижная фаза – вода с добавкой реактива А, метанол с добавкой реактива А, градиент от 5 до 40% метанола за 15 мин, скорость потока 2 мл/мин, детектор – УФ (254 нм): 1 – терефталевая кислота; 2 – ортофталевая кислота; 3 – изофталевая кислота
Необходимо, чтобы противоион растворялся в элюенте. Неправильный выбор противоиона может привести к образованию осадка, что вызовет возрастание значений k, размывание пика и заметное повышение давления на входе. Концентрация обычно колеблется от 0,01 М для противоиона с малой длиной цепи до 0,005 М для противоиона с более длинной цепью.
Для препаративных разделений ион-парную хроматографию не применяют, а количество вводимого образца сопоставимо с количествами, применяемыми для распределительной хроматографии. Увеличение максимально вводимого количества может быть достигнуто за счет предварительного образования ионных пар в образце. Для некоторых ионизированных (независимо от рН) анионов и катионов не требуется добавка буфера. Кислоты обычно разделяются при рН=4–7,4, а основания – при рН=2–5. При этом значения рН подвижной фазы могут для улучшения селективности разделения варьироваться.
Следует помнить, что ион-парная хроматография на обращенной фазе в целом метод более грубый, чем разделение на обращенной фазе, и должен использоваться, когда неприменимы распределительная хроматография на обращенной фазе или метод подавления ионов.
Литература
1. Baker D.R., George S. A/Amer. Lab., 1980, v. 12, No. 1, p. 41–46.
2. Bly D.D./Anal Chem, 1969, v. 41, No. 2, p. 477–480.
3. Drott E.E. //in Chromatographic Science Series, v. 8, Liquid Chromatography of Polymers and Related Materials, ed. J. Gazes. N.Y., M. Dekker, 1977, p. 41.
4. Krishen A., Tucker R.G./Anal. Chem., 1977, v. 49, No. 4, p. 898.
5. Mori S., Yamakctwa A./J. Liquid Chromatogr., 1980, v. 3, No. 3, p. 329 – 342.
6. Verzele M., Geeraert E./J. Chromatogr. Sci, 1980, v. 18, No. 10, p. 559 – 570.
7. Nettleton D.E./J. Liquid Chromatogr, 1981, suppl. No. 2, p. 359–398.
8. Rable F.M./International Lab, 1980, v. 10, No. 8, p. 91–98.
9. Small Bore Liquid Chromatography Columns //ed. R.P.W. Scott. N.Y., J. Wiley, 1984. 294 p.
10.Microcolumn High-Performance Liquid Chromatography //ed. P. Kucera. N. Y, Elsevier, 1984. 302 p.
11.Scott R.P.W., Kucera P./J. Chromatogr, 1979, v. 169, p. 51–62.
12.Scott R.P.W., Kucera P./J. Chromatogr, 1979, v. 185, p. 22–31.
13.Scott R.P.W./J. Chromatogr. Sci, 1980, v. 18, No. 1, p. 49–54.
14.Reese R.E., Scott R.P.W./J. Chromatogr. Sci, 1980, v. 18, No. 8, p. 479 – 486.
15.Scott R.P.W., Simpson C.F./J. Chromatogr. Sci, 1981, v. 19, No. 5, 224–233.