Исследование возможности извлечения редких металлов из золы-уноса ТЭЦ (MS Word 97)
СОДЕРЖАНИЕ: Министерство общего и профессионального образования Российской Федерации ДИПЛОМНАЯ РАБОТА Исследование возможности извлечения редких металлов из золы-уноса ТЭЦМинистерство общего и профессионального образования
Российской Федерации
ДИПЛОМНАЯ РАБОТА
Исследование возможности извлечения редких металлов из золы-уноса ТЭЦ
1999
СОДЕРЖАНИЕ
ВВЕДЕНИЕ............................................................................................................................................................................................ 3
1. ЛИТЕРАТУРНЫЙ ОБЗОР.............................................................................................................................................................. 4
1.1. Способы извлечения галлия из промышленных отходов............................................................................................. 4
1.1.1. Извлечение галлия из отходов алюминиевого производства.............................................................................. 5
1.1.2. Извлечение галлия из отходов переработки свинцово-цинковых руд............................................................ 12
1.1.3. Извлечение галлия из отходов переработки углей............................................................................................... 13
1.2. Способы извлечения ванадия из промышленных отходов........................................................................................ 19
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ........................................................................................................................................... 26
2.1. Характеристика золы............................................................................................................................................................ 26
2.2. Техника безопасности.......................................................................................................................................................... 27
2.3. Приборы и посуда.................................................................................................................................................................. 28
2.4. Реактивы................................................................................................................................................................................... 29
2.5. Методики анализов............................................................................................................................................................... 30
2.5.1. Фотометрическое определение железа с сульфосалициловой кислотой ..................................................... 30
2.5.2. Фотометрическое определение алюминия с алюминоном ............................................................................... 32
2.5.3. Фотометрическое определение ванадия ................................................................................................................ 35
2.5.4. Фотометрическое определение галлия ................................................................................................................... 37
2.5.5. Методика определения кремния ............................................................................................................................... 40
2.5.6. Выполнение сплавления и растворения навески силиката (золы) ................................................................. 42
2.6. Методики эксперимента....................................................................................................................................................... 43
2.6.1. Методика обескремнивания........................................................................................................................................ 43
2.6.2. Методика щелочной и кислотной обработки........................................................................................................ 43
2.6.3. Методика электрохимического выщелачивания.................................................................................................. 43
3. ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ....................................................................................................... 45
3.1. Отработка методики анализа на содержание ванадия............................................................................................... 45
3.2. Поведение галлия и ванадия при выщелачивании....................................................................................................... 45
3.2.1. Исследование влияния температуры и добавки NaCl на эффективность выщелачивания галлия, ванадия, железа и алюминия в сернокислой среде......................................................................................................................................... 46
3.2.2. Исследование эффективности выщелачивания галлия, ванадия, железа и алюминия щелочными растворами с добавкой NaCl............................................................................................................................................................................ 47
3.2.3. Исследование влияния концентрации щелочи, температуры и времени выщелачивания на полноту извлечения галлия........................................................................................................................................................................................... 48
3.2.4. Исследование возможности полного извлечения галлия при многостадийной обработке золы........... 50
3.3. Электрохимическое выщелачивание ванадия............................................................................................................... 50
3.3.1. Электровыщелачивание в кислой среде.................................................................................................................. 51
3.3.2. Электровыщелачивание в щелочной среде............................................................................................................ 51
ВЫВОДЫ.............................................................................................................................................................................................. 53
ЛИТЕРАТУРА...................................................................................................................................................................................... 54
ПРИЛОЖЕНИЯ................................................................................................................................................................................... 57
ВВЕДЕНИЕ
Роль редких металлов в современной технике все более возрастает. Редкие металлы в значительной мере определяют развитие таких важных отраслей промышленности, как производство специальных сталей, твердых и жаропрочных сплавов, электротехники, электровакуумной техники и ряда отраслей новой техники. За последние десятилетия новые металлы вошли в орбиту промышленного использования, возросли масштабы и расширилась номенклатура продуктов производств редких металлов.
Рассеянные редкие металлы встречаются обычно в виде изоморфных примесей в ничтожных концентрациях в кристаллах других минералов. Поэтому рентабельное извлечение рассеянных редких металлов возможно только из отходов производств основных металлов. Надо также учитывать и технологические трудности извлечения их из сырья.
В последнее время возрос интерес к извлечению редких металлов из нетрадиционных источников сырья, к которым следует отнести пыли и возгоны различных производств, в частности энергетики. Золу-уноса от сжигания угля можно рассматривать как техногенное сырье для получения многих ценных металлов, тем более, что она имеет пока нулевую стоимость. Поэтому извлечение редких металлов, наряду с основными компонентами (алюминий, железо), может сделать переработку золы-уноса рентабельной уже в ближайшем будущем.
Утилизация золошлаковых отходов ТЭЦ позволит расширить минерально-сырьевую базу, а также сократить земельные площади под золоотвалы и улучшить экологическую обстановку в зоне золоотвалов.
Целью нашей работы является поиск наиболее перспективных путей извлечения редких металлов из золы-уноса экибастузского угля.
1. ЛИТЕРАТУРНЫЙ ОБЗОР
1.1. Способы извлечения галлия из промышленных отходов
Галлий является одним из наиболее распространенных редких элементов. Среднее содержание его в земной коре составляет 1,510-3 вес. %. Самостоятельных минералов галлий не образует за исключением минерала галлита CuGaS2 , содержащего до 35% Ga [1].
Как основной компонент галлий не входит в минералы других элементов. Наиболее богатый источник галлия - очень редкий минерал германит Cu3 (Fe, Ge)S4 , содержащий 0,3 - 1,85% Ga.
Основная же масса галлия находится в рассеянном состоянии, накапливаясь, вследствие сходства химических и кристаллохимических свойств и близости размеров атомов и ионов галлия, алюминия и цинка, в виде изоморфной примеси в бокситах и сфалеритах. Галлий входит также в состав многих других минералов и руд, встречается в почвах, слюдах и морской воде [1].
Невысокая концентрация галлия в рудах не позволяет использовать их непосредственно в качестве сырья.
Галлий наряду с другими рассеянными элементами получают как побочный продукт при комплексной переработке тех руд, в которых он содержится в виде примеси. Источниками его получения являются промпродукты переработки медных, цинковых, алюминиевых, германиевых руд и углей, обогащенные галлием в силу отличительных свойств его соединений. Например, при переработке медных и цинковых руд, а также углей обогащению возгонов способствует высокая упругость халькогенидов галлия в состоянии низшей валентности; при переработке алюминиевых руд более прочные комплексные соединения галлия удерживаются и постепенно накапливаются в циркулирующих растворах глиноземного и содового производств и благодаря этому концентрируются относительно окиси алюминия [2].
Из-за отсутствия собственных руд промышленного значения объем производства галлия обусловлен масштабом переработки руд цветных металлов, используемых для его получения. При этом на долю продуктов и отходов производства глинозема и алюминия приходится более 90% всего добываемого количества галлия [3].
1.1.1. Извлечение галлия из отходов алюминиевого производства
Источниками получения галлия в алюминиевом производстве служат: оборотные алюминатные растворы, осадки последней карбонизации алюминиевых растворов, анодные сплавы после электролитического рафинирования алюминия, пыли электролизеров и угольные съемы от флотации электролитной пены. Содержание галлия в анодном сплаве и угольных съемах более или менее одинаково на всех алюминиевых заводах, и составляет около 0,2% в первом продукте и 0,05-0,07% во втором. Содержание же в оборотных алюминатных растворах и осадках карбонизации обусловлено количеством галлия в исходном сырье и технологией получения глинозема.
Наиболее перспективным источником получения галлия являются алюминатные растворы, содержащие галлаты. Из алюминатных растворов галлий получают двумя путями: 1) выделением из этих растворов галлиевого концентрата и затем из концентрата - металла; 2) электролизом растворов в ваннах с ртутным катодом, разложением амальгамы и выделением металлического галлия [1].
Галлиевые концентраты получают из обогащенных галлием гидратных осадков, образующихся при фракционной (стадийной) карбонизации алюминатных растворов, основанной на различных значениях величины pH осаждения гидроокисей галлия и алюминия.
Выделение галлиевого концентрата из оборотного алюминатного раствора при автоклавном способе может быть осуществлено по схеме: раствор алюмината подвергают двухстадийной карбонизации, при которой алюминий осаждается из раствора:
2NaAlO2 +CO2 +3H2 O=2Al(OH)3 +Na2 CO3
Первую карбонизацию проводят до тех пор, пока из раствора не выделится около 90% алюминия. При этом большая часть галлия остается в растворе. После повторной карбонизации фильтрата, при которой осаждается гидроокись алюминия, обогащенная галлием, содержание Ga2 O3 в осадке составляет 1-2% от содержания Al2 O3 [4].
По данным М. Бежа [5] процесс можно улучшить, сочетая выделение алюминия из растворов по способу выкручивания с двухстадийным выделением путем карбонизации. По способу выкручивания гидролиз алюмината натрия ускоряется введением в раствор затравки - гидроокиси алюминия:
![]()
![]() NaAlO2
+2H2
O Al(OH)3
+NaOH
NaAlO2
+2H2
O Al(OH)3
+NaOH
Часть алюминия (от 50 до 60%) первоначально выделяется выкручиванием. При этом осаждается незначительная часть галлия. Фильтрат затем обрабатывают по схеме двухстадийной карбонизации. При этом осадок первой (медленной) карбонизации, содержащий незначительную часть галлия, возвращается в оборот, что повышает общее извлечение галлия. Основная часть галлия выделяется из раствора в конце карбонизации вместе с последними фракциями гидроокиси алюминия и алюмокарбоната. Для извлечения галлия из гидратного осадка более богатых фракций разработано несколько методов концентрирования.
Кислотные методы получения галлиевых концентратов заключаются в обработке гидратных осадков соляной или серной кислотой и извлечении галлия из кислых растворов купферроном [6, 7] или экстракцией эфирами [3], или бутилацетатом [8].
По купферроновому методу гидратный осадок растворяют в серной кислоте, раствор фильтруют и подкисляют H2 SO4 до 1,5-2-н. концентрации. К раствору, охлажденному до 6-10°С, добавляют 6%-ный раствор купферрона. После длительного отстаивания объемистый осадок фильтруют и прокаливают при 600°С. При этом органическое соединение разрушается и остается смесь окислов. Алюминий не осаждается купферроном, однако захватывается осадком. Железо осаждается вместе с галлием. Для отделения железа прокаленный осадок может быть сплавлен с содой и сплав обработан водой. В раствор переходят галлий и алюминий, а железо остается в осадке.
При повторном осаждении галлия из раствора купферроном получается богатый галлием продукт, содержащий 30-50% Ga2 O3 .
Схема экстракционного метода получения галлия из гидратного осадка следующая. Галлий экстрагируется из кислых растворов эфирами или бутилацетатом. Экстракт галлия отделяют от раствора алюминия и взбалтывают с водой или со слабым раствором HCl. При этом хлорид галлия из органической фазы переходит в водную. Вместе с ним в водную фазу переходят некоторые металлы, которые осаждаются затем сероводородом в виде сульфидов. Если повторно использовать водный раствор хлорида галлия при реэкстракции его из органической фазы, то можно достичь высокой концентрации галлия в этом растворе. Выделить галлий из раствора его хлорида можно электролизом либо этого же раствора, либо раствора галлата натрия, полученного после осаждения гидроокиси из кислого раствора и растворения ее в щелочи.
Щелочные методы получения галлиевых концентратов основаны на растворении гидратных осадков в щелочи и последующей двухстадийной карбонизации полученного раствора. При второй стадии выделяется галлиевый концентрат с содержанием до 10% Ga2 O3 по отношению к окиси алюминия. Такая технология позволяет получать богатые галлием концентраты, но характеризуется низким извлечением металла, так как с гидроокисью алюминия первой стадии карбонизации теряется до 30-40% Ga2 O3 .
Наиболее эффективным и простым методом получения галлиевого концентрата из гидратных осадков является известково-карбонизационный [6, 9]. Гидратный осадок, репульпированный водой, обрабатывают сухой известью или известковым молоком, отделяют раствор галлата и алюмината натрия от алюмокальциевого осадка и затем выделяют галлий вместе с остатками алюминия при карбонизации раствора. Для получения металлического галлия галлиевый концентрат растворяют в горячей щелочи, очищают алюминатно-галлатный раствор от кремния известью и подвергают электролизу с выделением чернового металла [10].
Электрохимический метод основан на выделении галлия из алюминатных растворов электролизом на ртутном катоде [3]. Вследствие высокого перенапряжения водорода на ртути, кроме галлия, на катоде осаждаются многие другие элементы с отрицательными электродными потенциалами.
После насыщения галлием (0,3-1,0%) амальгаму промывают водой и разлагают раствором едкого натра при температуре, близкой к кипению, в герметическом реакторе в присутствии небольших кусочков (стружки) железа или графита. Разложение амальгамы может быть осуществлено также электрохимическим методом в системе амальгама (анод) - раствор NaOH - твердый катод.
В результате разложения амальгамы получается концентрированный раствор галлата натрия (содержит 10-80 г/л Ga) и ртуть, которую после некоторой, периодически осуществляемой очистки вновь применяют при электролизе. Из раствора галлата натрия электролизом в ваннах с неокисляющимся катодом (из специальной стали или жидкого галлия) выделяют металлический галлий. Однако он загрязнен примесями цинка, свинца, меди и др.
Иногда с целью получения более чистого галлия галлатные растворы перед электролизом обрабатывают сероводородом для осаждения примесей тяжелых цветных металлов в виде сульфидов.
Ванадаты, молибдаты, сульфаты и фосфаты натрия, присутствующие в алюминатных растворах в значительных количествах (0,1-0,8 г/л), восстанавливаются на ртутном катоде до нерастворимых в щелочи соединений низших валентностей и переходят в осадок, образуя ванадиево-молибденовый концентрат.
Галлий можно извлечь из алюминатных растворов цементацией амальгамой натрия. Скорость цементации галлия натриевой амальгамой зависит от температуры, концентрации натрия в амальгаме и контакта реагирующих сред.
А.Т. Нижник и З.В. Шехтер установили, что из раствора, содержащего 50 г/л NaOH и 0,4 г/л Ga можно извлечь около 90% галлия, если этот раствор перемешивать с 1%-ной амальгамой натрия со скоростью 400 об/мин при температуре 50°С [11]. Наличие примесей в щелочных галлиевых растворах не оказывают заметного влияния на скорость и глубину цементации галлия амальгамой натрия. Лишь в присутствии ванадия скорость цементации заметно снижается. Примеси, разлагая амальгаму, увеличивают ее расход, однако алюминий и пятивалентный мышьяк не восстанавливаются натриевой амальгамой и не влияют на ее расход. Цинк, аналогично галлию, восстанавливается до металла и переходит на катод.
Можно также извлекать галлий из алюминатных растворов цементацией на галламе алюминия [12]. Восстановление галлат-аниона алюминием, растворенным в галлии, идет по реакции:
![]()
![]() NaGaO2
+2Al+NaOH+H2
O Ga+2NaAlO2
+
NaGaO2
+2Al+NaOH+H2
O Ga+2NaAlO2
+![]() H2
H2
Использование галламы алюминия, в отличие от амальгамы имеет преимущество в том, что при этом не существует ограничения растворимости галлия, процесс нетоксичен, величина перенапряжения водорода на галламе алюминия выше, чем на твердом алюминии, что улучшает условия выделения галлия. Выделение галлия цементацией на галламе алюминия целесообразно применять к растворам, предварительно очищенным от ванадат-ионов, так как восстановление последних до низших ступеней валентности снижает степень цементации галлия с 90-93 до 40-47%.
В 1966 году опубликован способ [13] осаждения галлия из раствора алюмината натрия активным алюминиевым порошком, взятым в большом избытке. Реакция протекала при разности потенциалов систем Al/Al3+ 1,67в, Ga/Ga3+ 0,52в
![]()
![]() Ga(OH)`
4
+Al Al(OH)`
4
+Ga
Ga(OH)`
4
+Al Al(OH)`
4
+Ga
Полученный осадок сплава содержал до 80% Ga. Сплав хлорировали при температуре более 300°С, получали смесь хлоридов, из которой извлекали жидкостной экстракцией алифатическими и гидроароматическими углеводородами.
Имеются сведения [3] об электролитическом осаждении галлия из алюминатных растворов на твердых катодах из свинца и меди. Катоды представляют собой пластины большой площади или движущуюся ленту. Электролиз проводился при катодной плотности тока 4-8 А/дм2 , температуре 40-50°С и напряжении на ванне 5-6в. В этих условиях галий выделялся на катоде вместе с другими примесями (цинком, железом) в виде сплава. Выход галлия по току составлял 0,1-1,0%. В начале электролиза, когда поверхность катода была чистой и концентрация галлия в растворе была максимальной (около 0,2 г/л), он осаждался вполне удовлетворительно. Однако вследствие пассивирования катода, которое наступало довольно быстро, выделение снижалось и затем прекращалось совсем. В результате воздействия цинка, органических примесей, молибдена, ванадия и железа на катоде образовывался черный налет. Чтобы удалить его и обновить поверхность катода, его периодически обрабатывали в течение 5-20 мин горячей концентрированной щелочью. Галлий и другие примеси переходили в щелочной раствор; концентрации галлия в нем достигали 5-50 г/л, то есть достаточно для выделения его в виде металла на катоде из нержавеющей стали.
Для извлечения галлия из анодного сплава – остатка, образующегося при электролитическом рафинировании алюминия, – применяют щелочные и кислотные способы.
Щелочной способ вскрытия сплава состоит в обработке измельченного материала горячим раствором едкого натра. При этом алюминий и галлий переходят в раствор в виде алюмината и галлата натрия, железо остается в нерастворимом остатке, кремний распределяется между раствором и остатком. Реакция растворения протекает бурно с выделением водорода и разогревом.
Щелочное вскрытие сплава позволяет отделить галлий и алюминий от меди и железа, что упрощает дальнейшее извлечение галлия, которое можно осуществить одним из известных способов извлечения его из алюминатных растворов. Щелочной способ разложения анодных сплавов дает удовлетворительное извлечение только в применении к сплавам, содержащим 25-30% алюминия [14].
Для переработки бедных алюминием отработанных анодных сплавов, получаемых в последнее время, пригодны только кислотные методы. Разлагать сплав можно как выщелачиванием измельченного сплава серной или соляной кислотой, так и анодным растворением [2]. В раствор наряду с галлием и алюминием переходят также железо и частично (за счет окисления кислородом воздуха) медь. Так как железо осаждается купферроном, в этом случае применять купферрон невыгодно, и перерабатывают растворы экстракционным путем, используя бутилацетат или трибутилфосфат. Если разложение велось серной кислотой, к раствору добавляется соответствующее количество хлорида натрия. Чтобы отделить железо, раствор перед экстракцией обрабатывают каким-либо восстановителем, например железной стружкой. Для реэкстракции галлия из органического слоя последний промывают водой. После экстракции следует очистка от примесей молибдена и олова осаждением сернистым натрием и, наконец, электролиз щелочного раствора галлата с целью получения металлического галлия.
Гастингер [15] разработал технологическую схему извлечения галлия из анодного сплава, в основу которой положил солянокислый способ вскрытия сплава и хлоридный метод разделения галлия и алюминия в растворе, насыщенном хлористым водородом, где хлориды этих металлов имеют различную растворимость.
Растворением измельченного анодного сплава в соляной кислоте достигалось отделение меди, которая оставалась в шламе. Для отделения галлия от цинка и меди из солянокислого раствора осаждали аммиаком гидроокиси галлия и железа, при этом цинк и медь оставались в растворе в форме аммиакатов. Для отделения железа использовали тиосульфат натрия.
Разработана также технологическая схема извлечения галлия из сплава, в основу которой положен метод адсорбции галлия на активной двуокиси марганца [6]. В методе адсорбции вскрытие сплава проводится электролитическим растворением в серной кислоте с последующим извлечением галлия из раствора адсорбцией на активной двуокиси марганца. Десорбция галлия осуществляется обработкой осадка 10-12%-ным раствором щелочи.
1.1.2. Извлечение галлия из отходов переработки свинцово-цинковых руд
При получении из полиметаллических сульфидных свинцово-цинковых руд тяжелых цветных металлов галлий извлекается попутно с другими редкими и рассеянными элементами – кадмием, германием, индием, таллием, рением и др. [1].
В цинковом производстве наиболее богаты галлием цинковые и свинцовые кеки и ретортные остатки. Продукты свинцового производства содержат значительно меньше галлия; получение его может рассматриваться только как попутное при извлечении других редких и рассеянных элементов. Одним из рациональных способов извлечения галлия из щелочных растворов, получающихся при переработке отходов свинцово-цинкового производства, с одновременным отделением его от ряда примесей, является сульфидный, основанный на соосаждении сульфида галлия с сульфидом цинка при обработке этих растворов сульфидом натрия [16].
Продукты цинкового производства, обогащенные галлием, выщелачивают серной кислотой; при этом галлий, цинк и железо растворяются, свинец остается в шламе. После отделения шлама раствор нейтрализуют окисью цинка до pH=5 с целью выделения в осадок гидроокисей галлия и железа. Растворением осадка в щелочи разделяют галлий и железо. Многократное повторение операций растворения и выделения осадка дает возможность получать концентраты, содержащие до 10% Ga2 O3 . Концентрат растворяют в щелочи и извлекают галлий электролизом.
1.1.3. Извлечение галлия из отходов переработки углей
Галлий постоянно присутствует в углях, но только в незначительных количествах. Он содержится как в углеродной массе, так и в минералогической составляющей, в которой он изоморфно замещает алюминий в его окиси. Форма вхождения галлия в угли не исследована, однако на основании его свойств можно предположить, что это могут быть адсорбированные сульфиды, карбонаты, хлориды, а также сложные карбиды и карбонилы.
Летучесть некоторых соединений галлия с серой, кислородом, хлором и углеродом определяет возможность концентрирования галлия в летучих возгонах процессов сжигания, газификации, полукоксования и коксования углей. Однако вхождение галлия преимущественно в минералогическую составляющую затрудняет этот процесс. При использовании углей в качестве энергетического топлива образующиеся шлаки в большинстве случаев содержат лишь следы галлия. Летучие золы в системе дымоходов несколько богаче галлием по сравнению с углями. Содержание галлия в возгонах ограничивается 0,1% и обусловлено летучестью низших окислов и сульфидов. Сжигание при недостатке кислорода сопровождается большей степенью возгонки; при сжигании углей в сильно окислительной атмосфере галлий концентрируется в шлаках. Известно несколько способов получения галлия и германия из пылей и сажистых уносов, из золы, а также других побочных продуктов переработки каменного угля, в частности, из промывных вод смолоотделительных систем.
Способ Моргана и Девиса [17] заключается в том, что пыль газовых заводов обрабатывали достаточно концентрированной соляной кислотой, затем отгоняли тетрахлорид германия и из маточного кислотного остатка после доведения кислотности до 6-н. и восстановления железа до двухвалентного извлекали галлий экстракцией диэтиловым эфиром. После отгонки эфира хлориды переводили в сульфаты, разбавляли водой и осаждали гидроокись галлия, которую затем переосаждали для отделения железа и других примесей.
Исследователи В.М. Кострикин и Б.Н. Иванов-Эмин в качестве сырья применяли озоленные сажистые уносы одной из газостанций [18].
Эту золу обрабатывали в дистилляционных аппаратах соляной кислотой при температуре 150°С. Образующийся тетрахлорид германия отгоняли вместе с парами соляной кислоты и направляли на получение германия. Кислотные остатки промывали водой при кипячении в течение 30 мин. Нерастворимый остаток, состоящий в основном из гидрата окиси кремния, отфильтровывали.
Для отделения алюминия и галлия от железа последний восстанавливали тиосульфатом натрия, затем из раствора с помощью анилина осаждали гидроокиси галлия и алюминия. Полученные осадки содержали: алюминий, серу, титан, ванадий, железо и практически весь галлий. Осадки отфильтровывали, промывали водой или 3%-ным раствором сульфата алюминия, затем обрабатывали 6-н. соляной кислотой, отфильтровывали от серы. Галлий из раствора выделяли с помощью ферроцианида калия, для чего раствор нагревали до кипения. Для осаждения использовали 10%-ный раствор осадителя. Осадок отфильтровывали, промывали слабым раствором ферроцианида калия и озоляли.
Полученные полуторные окислы содержали от 0,3 до 0,75% окиси галлия. Окиси сплавляли с перекисью натрия, сплав выщелачивали водой. Раствор, содержащий галлий и алюминий, нейтрализовывали аммиаком до слабокислой реакции. Гидроокиси растворяли в соляной кислоте и при 6-н. кислотности галлий извлекали эфиром. Эфир отгоняли, остаток растворяли в слабой соляной кислоте. Из раствора осаждали гидрат окиси галлия.
Разработан промышленный процесс переработки возгонов газификации углей.
Первой операцией является удаление кремния и окиси алюминия, что достигают плавлением с окисью меди и определенным флюсом. В результате получают шлак, который удаляют, и металлический штейн, содержащий медь, железо, мышьяк, серу, германий и галлий. Штейн растворяют в разбавленном растворе хлорида железа при пропускании хлора. Полученный раствор переносят в сосуд для дистилляции и после добавления серной кислоты до 7-н. кислотности производят дистилляцию и конденсацию паров в две фракции. Первую фракцию - раствор AsCl3 в соляной кислоте (1:1) – отбрасывают, вторую фракцию, содержащую тетрахлорид германия и до 20% AsCl3 , используют для выделения германия; галлий извлекают из кубового остатка.
Этот раствор направляют на выпарной аппарат и обрабатывают алюминиевыми стружками для осаждения меди и других тяжелых металлов и восстановления железа. Отфильтрованный раствор доводят до 7-н. кислотности по соляной кислоте и экстрагируют изопропиловым эфиром. Хлорид галлия, растворенный в эфире, извлекают 2%-ным раствором соляной кислоты; эфир после дистилляции используют повторно. Раствор хлорида галлия обрабатывают сероводородом для отделения молибдена и мышьяка, которые растворяются в эфире. Раствор после отделения осадка фильтрованием кипятят для удаления сероводорода. После окисления азотной кислотой раствор обрабатывают избытком раствора едкого натра и направляют на электролиз.
В последние годы появились обзорные и технологические работы японских [19], польских, чешских, венгерских и других исследователей по извлечению галлия и германия из летучих возгонов, полученных в процессе сжигания и газификации углей, а также промывных вод отделителей смолы. Для возгонов особое значение приобрели щелочные способы переработки, которые заключаются в сплавлении или спекании их с едким натром или с содой [19], окисью меди и углем и спекании с известняком. Последним из способов можно получить также и глинозем.
Нейтрализация спека или плава [20] соляной кислотой до концентрации 0,2-н. HCl позволяет выделить в осадок основную часть гидрата окиси алюминия и кремния. После отделения осадка раствор нейтрализуют до pH=5 и из него осаждают гидраты окиси галлия и германия. Осадок редких металлов растворяют в соляной кислоте и из раствора германий осаждают в виде дисульфида GeS2 (при концентрации 4-н. HCl) или германата магния. Галлий выделяется из раствора в виде гидроокиси.
Щелочные растворы галлата, германата и алюмината можно перевести в солянокислые и оттуда галлий извлечь экстракцией. Эфирный экстракт обрабатывают раствором едкого натра и электролизом выделяют металлический галлий. Извлечение галлия из золы по этой схеме составляет 80%.
По схеме извлечения галлия и глинозема методом спекания золы и пыли с известняком [21] золу или другие углистые продукты смешивают с известняком и спекают при 1325-1375°С. В процессе спекания окись галлия, аналогично окиси алюминия образует с известью галлаты кальция составов 5CaO3Ga2 O3 и CaOGa2 O3 . Спек выщелачивают раствором соды, при этом алюминий и галлий переходят в раствор, а кремнезем в виде двухкальциевого силиката вместе с гидратом окиси железа остается в шламе. В результате выщелачивания в алюминатный раствор извлекается из золы 90-93% галлия. Получить его из этого раствора можно карбонизационно-известковым способом.
Для использования летучих возгонов необходимо обогащать их редкими металлами до концентраций, окупающих их дальнейшую переработку. Для переработки золы предлагают восстановление ее при температуре 1000°C с отгонкой редких элементов в пыль, восстановление водородом при температуре более 500°C с отгонкой, сублимацию содержащихся соединений при температуре более 700°С в атмосфере CO2 ; CO2 +CO, Ar и др. при пониженном давлении. Для дополнительного обогащения золы предлагают применять восстановительный обжиг при температуре более 1000°C и использовать в качестве восстановителя C, CO, CH4 или H2 .
При сжигании угля галлий в большей степени переходит в возгоны и теряется с дымовыми газами. Японские исследователи предложили промывать эти отходящие газы щелочными и кислотными растворами и охлаждать. При этом галлий переходит в раствор, из которого после удаления сажи и нерастворимых примесей его вместе с железом и германием осаждают танином.
Другой метод извлечения галлия из воды заключается в следующем. Газовую воду после удаления аммиака смешивают с неочищенной водой до pH=89 и раствор окисляют воздухом, который продувают в течение 2-3 ч при 75-85°C, а затем подкисляют серной кислотой до pH=23. При этом получают смолистый органический осадок, содержащий основное количество галлия и германия, из которого после озоления и обработки концентрированной соляной кислотой германий извлекают в виде его тетрахлорида, а галлий экстракцией изопропиловым эфиром с последующим осаждением гидроокиси, растворением её в щелочи и электролизом.
Известен способ получения галлия из воды от промывки газа или орошения возгонов. Воду упаривают до ![]() первоначального объема и эфиром извлекают фенолы.
первоначального объема и эфиром извлекают фенолы.
При дальнейшем упаривании получают органическую массу, в которой содержатся редкие металлы. Её озоляют при 600-700°C и сплавляют с едким натром, сплав выщелачивают соляной кислотой, из солянокислого раствора отгоняют германий, а из остатка экстрагируют галлий.
Один из способов извлечения галлия и германия из побочных продуктов переработки каменных углей заключается в следующем. Возгоны промывают кислыми и щелочными растворами, эти растворы окисляют, к ним добавляют Fe(II), Fe(III), Ni, Cu или Al в виде солей в трехкратном отношении к германию и кремнию и соосаждают галлий и германий при pH=67,3, если добавлены Fe(II), Cu или Ni и при pH=4,56, если добавлены Fe(III) или Al.
В другом варианте растворы, содержащие галлий, окисляют воздухом, озоном, перекисью водорода, KMnO4 , K2 Cr2 O7 при pH раствора более 6. В результате соединения галлия и германия переходят в осадок. Этот осадок сушат, прокаливают при 400-450°C и получают золу. Золу обрабатывают HCl и Cl2 , затем германий удаляют дистилляцией, остаток фильтруют, экстрагируют и из реэкстракта извлекают галлий.
Польские исследователи предлагают из растворов с концентрацией галлия и германия не более 0,01 мгл-1 выделять их путем хемосорбции гумминовыми соединениями, поглощающая способность которых зависит от pH и поэтому условия адсорбции могут быть подобраны для каждого из элементов.
Разработана [22] кислотно-экстракционная технология извлечения галлия из золы-уноса от сжигания энергетических углей. В работе использовали зольные уносы от сжигания экибастузского угля следующего состава, %: Al2 O3 – 15,65; Fe2 O3 – 17,24; CaO – 10,82; Mg – 4,98; SiO2 – 28,50; Zn – 4,33; Na2 O – 2,15; K2 O – 2,27; Ga – 0,019 [23]. Золу обрабатывали соляной кислотой концентрацией 4 моль/дм3 при температуре 80°C, продолжительности 2 ч, соотношении Т:Ж=1:4. В раствор переходит до 85% галлия, кремний практически весь остается в нерастворимом остатке. Из солянокислого раствора галлий экстрагировали 0,3-М раствором триалкиламина в керосине с добавлением 20% эксола. Экстракцию вели в противотоке в 6 ступенях экстрактора ящичного типа. При этом галлий практически полностью перешел в органическую фазу (99,6%). Реэкстракцию проводили раствором NaOH с pH=12,54. Извлечение галлия по предложенной схеме составило 76-77%.
1.2. Способы извлечения ванадия из промышленных отходов
Ванадий широко распространен в природе и составляет около 0,02% от веса земной коры, то есть примерно столько же, сколько цинк и никель. Однако он более рассеян и присутствует виде следов во многих рудах. Месторождения собственно ванадиевых руд в природе встречаются довольно редко. Небольшое его количество найдено в железных, свинцовых, свинцово-цинковых, свинцово-медных и алюминиевых рудах [24]. Практически весь ванадий земной коры находится в её твердой оболочке – литосфере – в изверженных породах, где он вследствие близости размеров ионных радиусов V3+ и Fe3+ изоморфно замещает катион железа (III) [25].
В связи с отсутствием собственных руд ванадия, рентабельных для эксплуатации, этот элемент выделяют из различных полупродуктов. Ванадий добывают из отходов переработки карнотитовых руд (сырье для получения урана) и в небольших количествах – при переработке апатитов и бокситов. Самыми важными источниками ванадия являются некоторые асфальты, в которых его содержание доходит до 25%, битуминозные сланцы и нефть. Из других важных источников получения ванадия следует назвать шлаки, получающиеся при переработке титаномагнетитовых руд. В Польше небольшие количества ванадия извлекают из шлаков медеплавильных заводов [26].
Ванадий является исключительно литофильным элементом и при флотации медных руд переходит в концентрат в количестве около 50%. При выплавке медного штейна ванадий из-за своей литофильности концентрируется в шлаке.
Аналогично и в процессе выплавки чугуна из руд, содержащих ванадий, этот элемент переходит в шлак. Причем особо богаты ванадием конвертерные шлаки.
Ванадий в шлаках содержится в виде соединений типа шпинели FeOV2 O3 и MnOV2 O5 . В мартеновских шлаках ванадий, кроме того, входит в состав силикатов типа оливина (Mg, Fe)2 SiO4 и пироксена CaFeSi2 O6 [27]. Ванадиевые шлаки представляют собой ванадиевые концентраты, относительно легко перерабатываемые на V2 O5 или ванадат кальция.
Переработка ванадиевых шлаков наиболее эффективно производится следующими способами: 1) окислительным обжигом с поваренной солью или сильвинитом; 2) окислительным обжигом с содой; 3) хлорированием. Наиболее пригодно для передельных шлаков вскрытие путем окислительного обжига с поваренной солью. Из шлаков, содержащих более 12% CaO, обжиг с содой дает более высокое извлечение, чем обжиг с поваренной солью. Хлорированием извлекают из конвертерных шлаков наряду с ванадием также и титан.
Механизм обжига шлаков с NaCl изучен М.Н. Соболевым с сотрудниками [25]. Показано, что выше 800-850°C в окислительной атмосфере реакция
2NaCl+O2 =Na2 O+Cl2
значительно ускоряется в присутствии окислов железа, марганца и особенно V2 O5 . Образующаяся Na2 O реагирует с V2 O5 :
Na2 O+V2 O5 =2NaVO3
Окислительная атмосфера в зоне обжига способствует окислению V(III), входящего в состав шпинели. Выделяющийся при обжиге хлор также участвует в процессе вскрытия шпинели:
FeOV2 O3 +4Cl2 =2VOCl3 +FeCl3 +O2
Хлорированию подвергают смесь измельченного шлака со стехиометрическим по отношению к ванадию количеством NaCl. Спек после обжига охлаждают и выщелачивают водой. В раствор переходит до 95% ванадия. Содержание ванадия в растворе достигает 30 г/дм3 в расчете на V2 O5 . Из осветленного раствора добавкой серной кислоты осаждают V2 O5 xH2 O, который затем сушат и прокаливают до V2 O5 .
После выделения около 96% V2 O5 остаток ванадия из раствора высаживают добавкой известкового молока, получая труднорастворимый 2CaOV2 O5 Ca(OH)2 . Полученный осадок используют в производстве феррованадия.
При переработке бокситов методом Байера примерно 65% ванадия переходит в шлам, остальная часть находится в растворе. Увеличению доли ванадия, переходящего в алюминиевый раствор, благоприятствует его пятивалентная форма. При наличии в алюминатном растворе 100-300 г/л Na2 O растворимость солей ванадия можно значительно снизить, добавив NaF в соотношении V2 O5 :NaF=1:2. Ванадий переходит в осадок в виде соли 2Na3 VO4 NaF19H2 O [28]. По другим данным, для переработки ванадиевого концентрата, являющегося побочным продуктом глиноземного производства, предложена следующая технологическая схема. Ванадиевый концентрат (до 38% V2 O5 и 60% влаги) растворяют в 20%-ном растворе NaOH. В раствор извлекается 99,4% V. После фильтрации к раствору добавляют NH4 Cl и выделяют NH4 VO3 , который очищают перекристаллизацией. Соль сушат и прокаливают при 500-550°C. Выход V2 O5 – 94% [29].
Отходы уранового производства, содержащие ванадий, – ценное сырье. Перспективен метод их переработки хлорированием в присутствии восстановителя. Продукт хлорирования VOCl3 – легколетучая жидкость, которую подвергают дистилляционной очистке, гидролизу в присутствии аммиака, последующей сушке и прокалке до V2 O5 .
В практике работы промышленных предприятий для выделения ванадия из растворов, получаемых после переработки карнотитовых руд, применяются ионитные и экстракционные процессы. В современной металлургической промышленности сорбционные методы получают все большее распространение [30].
Существует способ переработки патронитовых ванадийсодержащих концентратов, полученных из зол асфальтитов, заключающийся в многократной обработке руд крепкой щелочью и переводом ванадия в раствор. Для нейтрализации растворов используют азотную кислоту. Ванадий после выпаривания и разбавления растворов осаждают хлористым алюминием.
В ряде патентов предлагается обработка ванадийсодержащих материалов растворами сульфида натрия или проведение плавки с применением в качестве шихтовых материалов смеси сульфата натрия с углеродом. При этом образуются сульфованадаты натрия, которые легко растворяются в воде. Осторожным подкислением выделяется ванадий в форме сульфидов (V2 S5 или V2 S3 ). В данных процессах достигается очень хорошее отделение ванадия от железа [31, 32].
Разработан процесс извлечения некоторых металлов (Li, B, V и Ga) из летучей угольной золы [33]. Процесс включает 3 операции: двухстадийное выщелачивание серной кислотой при двух различных концентрациях, концентрирование редких металлов из раствора с применением хелатных смол и очистка каждого металла экстракцией. Галлий и ванадий выщелачивают 3-н. раствором H2 SO4 , затем концентрируют их совместно на хелатной смоле иминодиацетатного типа и очищают экстракцией с применением триоктиламмонийхлорида и ди(2-этилгексил)фосфорной кислоты соответственно.
Другой кислотный метод обработки золы был запатентован в США [34]. Он заключается в выщелачивании золы, содержащей V и Ni, соляной кислотой с промотором растворения (V2 O5 , гипохлориты натрия, калия, кальция, пероксиды, ClO2 ). Затем щелока отделяют от остатка и обрабатывают гидроксидом натрия, калия или кальция, повышая pH раствора до уровня 5,5-6,5(6,2). Выпадающий при этом осадок, содержащий соединения V(3+) и V(4+), отделяют от раствора, который затем дополнительно подщелачивают (до pH 8,5-9,5), выделяя в осадок гидроксид никеля. Ванадийсодержащий осадок высушивают, после чего смешивают с гидроксидом натрия, калия или кальция, смесь прокаливают на воздухе при 500-1000°C (950°C), в результате чего ванадий окисляется до пятивалентного состояния. V(5+) выщелачивают водой, полученные щелока подкисляют соляной кислотой, выделяя в осадок V2 O5 .
Предложены также щелочные способы обработки золы с целью извлечения ванадия. В одном из способов [35] ванадийсодержащую золу подвергают гравитационному разделению. Обогащенную ванадием фракцию выщелачивают щелочным раствором, продувая одновременно газ-окислитель (например, сжатый воздух). Щелочной раствор концентрируют, затем пропускают через него CO2 -содержащий газ до pH 8-9. Осадок отделяют. К раствору добавляют соль аммония (NH4 Cl) для извлечения ванадия в виде аммонийной соли.
В другом способе [36] смоченную золу-уноса подвергают разделению для образования тонкодисперсного углеродного продукта и не содержащей углерода водной суспензии. Полученную суспензию выщелачивают при повышенном давлении в растворе гидроксида щелочного металла с концентрацией менее 5 моль/л при 110-300°C. Суспензию разделяют на жидкую и твердую фазы для получения щелока от выщелачивания, обогащенного ванадием, и обедненного ванадием остатка. Щелок от выщелачивания приводят в контакт с экстрагентом, содержащим четвертичный амин и производное оксима. Затем насыщенный экстрагент приводят в контакт с водным раствором для получения ванадийсодержащего водного щелока и регенерированного экстрагента. Из ванадийсодержащего щелока извлекают ванадийсодержащие соединения.
Предложены способы извлечения ванадия из ванадийсодержащего сырья путем электрохимического выщелачивания [37, 38]. В этих исследованиях в качестве электролита предлагают использовать растворы, содержащие хлориды и карбонаты щелочных металлов. Электролиты, содержащие хлор-ионы, вызывают коррозию аппаратуры, использование карбонатов сопряжено с трудностями регенерации отработанного электролита. Объектом служил ванадийсодержащий конвертерный шлак Нижне-Тагильского металлургического комбината с содержанием пятиокиси ванадия 18,38%. Задача поисковых исследований заключалась в нахождении оптимальных параметров процесса, подборе конструкции и вида материалов электролизёров и электродов, состава электролитов. Факторы: температура и время выдерживания, плотность тока, концентрация электролита, высота и навеска шлакового слоя, перемешивание, введение активирующих веществ, отношение Т:Ж, а также температура и продолжительность предварительной термической обработки шлака. Были опробованы различные конструкции электролизёров и электродов из графита, никеля, свинца и нержавеющей стали. Наиболее подходящими оказались электроды из нержавеющей стали. Электролизёр имел форму стакана из оргстекла, катод представлял собой полый цилиндр, через который проходило перемешивающее устройство. Вокруг катода располагались аноды, сделанные в виде лопастей. Нижняя часть катода отделялась от среды фильтровальным материалом.
При использовании в качестве электролита серной кислоты (5-13%) степень перехода ванадия в раствор составила в пределах 60-80%. При этом раствор в значительном количестве загрязнялся железом, алюминием, фосфором и другими примесями. Так же не дали желаемого результата электрохимическое выщелачивание шлака в растворе едкого натра, хлорида натрия и их смеси. Извлечение ванадия в растворе не превышало 30-40%. Поэтому дальнейшие исследования проводили с предварительно обожженным шлаком. В качестве электролита использовали раствор едкого натра различной концентрации. Видимо, предварительная термическая обработка шлака (780-800°C, 15-20 мин) способствует разрушению его силикатной составляющей вследствие окисления шпинелида, что интенсифицирует процесс электрохимического выщелачивания.
Можно предположить, что увеличение скорости окисления ванадия низких валентностей происходит и за счет выделения атомарного кислорода при электрохимическом процессе.
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
2.1. Характеристика золы
Зола и шлак ТЭЦ представляет собой остаток от сжигания твердого топлива. Они являются продуктами высокотемпературной (до 1200-1700°C) обработки минеральной, несгорающей части углей. При этом в камерных топках получают отходы двух видов: зола-уноса и шлак.
Шлак образуется в результате сжигания размягченных частиц золы в объеме топки или на ее стенках и накапливается в шлаковом бункере под топкой. Размер зерен шлака 150 мм. Зола-уноса уносится из топки с дымовыми газами и улавливается при их очистке в циклонах и электрофильтрах. Размер частиц золы менее 1 мм. Свыше 80% минеральной части углей переходит в золу, до 20% – в шлак.
Химический состав золы ТЭЦ-4
(экибастузский уголь)
| SiO2 | Al2 O3 | FeO | CaO | MgO | SO2 | TiO2 | K2 O | Na2 O | P2 O5 | MnO2 |
| 61-62% | 27,3% | 5,65% | 1,17% | 0,49% | 0,52% | 1,49% | 0,42% | 0,32% | 0,52% | 0,17% |
Содержание химических элементов в золе, % масс.
| Элементы | % масс. |
| Кремний, (Si) | 29 |
| Железо, (Fe) | 4,0 |
| Кальций, (Ca) | 0,52 |
| Алюминий, (Al) | 11,0 |
| Магний, (Mg) | 0,16 |
| Стронций, (Sr) | 0,044 |
| Титан, (Ti) | 0,38 |
| Марганец, (Mn) | 0,082 |
| Барий, (Ba) | 0,20 |
| Иттрий, (Y) | 0,0040 |
| Лантан, (La) | 0,0014 |
| Церий, (Ce) | 0,0066 |
| Иттербий, (Yb) | 0,0006 |
| Тербий, (Tb) | 0,0008 |
| Диспрозий, (Dy) | 0,0009 |
| Самарий, (Sm) | 0,0005 |
| Торий, (Th) | 0,0006 |
| Уран, (U) | 0,0002 |
| Цирконий, (Zr) | 0,034 |
| Медь, (Cu) | 0.0056 |
| Ванадий, (V) | 0,014 |
| Галлий, (Ga) | 0,0044 |
Примечание: анализ золы проводился в институте Гидроцветмет (г. Новосибирск).
2.2. Техника безопасности
Техника безопасности при выполнении любой работы в химической лаборатории должна быть предметом постоянного внимания, так как даже незначительная неосторожность и невнимательность могут привести к несчастным случаям с тяжелыми последствиями.
Правила работы с электрическим оборудованием.
При работе с электрооборудованием необходимо соблюдать следующие требования:
· электрооборудование должно быть исправным;
· при работе с электрооборудованием, находящимся под напряжением, применять исправные средства защиты (резиновые перчатки, изолирующие подставки и др.), работу проводить инструментом с изолированными рукоятками;
· в случае перерыва подачи электроэнергии все приборы должны быть немедленно выключены;
· в случае загорания проводов или электрооборудования, находящегося под напряжением, необходимо выключить электроэнергию и тушить огонь сухим углекислотным огнетушителем, сухим песком, покрывалом из асбеста.
Правила работы с едкими веществами.
Едкие вещества (кислоты, щелочи), попадая на кожу, вызывают ожоги. Особая опасность заключается в возможности поражения глаз. Поэтому необходимо соблюдать следующие условия:
· при любых работах с едкими веществами обязательно применение защитных очков или масок;
· переливать кислоты только при включенной тяге в вытяжном шкафу;
· растворение щелочей необходимо производить прибавлением к воде небольших кусочков, куски щелочи брать только щипцами;
· кислоты следует разбавлять в термостойких стаканах прибавлением их к воде небольшими порциями, разлитые кислоты и щелочи следует немедленно нейтрализовать и только после этого проводить уборку
В лаборатории для оказания первой помощи должна быть аптечка, а также растворы борной кислоты, бикарбоната натрия и перманганата калия.
2.3. Приборы и посуда
1. Колориметр фотоэлектрический концентрационный КФК-2 №838440. Рабочий диапазон длин волн 315-980 нм. Пределы измерения оптической плотности от 0 до 1,5.
2. Термостат ЕЗЕ №3178.
3. Мешалка механическая РМ-70/30, рабочее напряжение 220В, сила тока 0,2A, частота 30Гц, максимальное число оборотов 3000 в мин.
4. Весы аналитические А-3/200. Точность измерения 10-4 г, заводской номер 133051.
5. Весы технические. Точность измерения 10-1 г.
6. Плитка электрическая, рабочее напряжение 220 В.
7. Муфельная печь ПМ-8 №42048. Напряжение 220 В, ток переменный, максимальная температура 1100°C, частота 50 Гц, мощность 1,8 кВт.
8. Стаканы термостойкие вместимостью 100, 250, 400, 1000 см3 .
9. Колбы мерные вместимостью 50, 100, 200, 250, 1000 см3 .
10. Пипетки мерные вместимостью 5, 10, 20, 25 см3 .
11. Цилиндры мерные вместимостью 10, 100 см3 .
12. Колбы плоскодонные термостойкие вместимостью 100 см3 .
13. Фарфоровая ступка.
14. Фарфоровые тигли.
2.4. Реактивы
1. Соляная кислота HCl, х.ч.
2. Азотная кислота HNO3 , х.ч.
3. Серная кислота H2 SO4 , х.ч.
4. Ортофосфорная кислота H3 PO4 , х.ч.
5. Кислота сульфосалициловая 2-водная C7 H6 O6 S2H2 O, х.ч.
6. Аммиак NH3 , х.ч.
7. Ледяная уксусная кислота CH3 COOH, х.ч.
8. Гидроокись натрия NaOH, х.ч.
9. Железоаммонийные квасцы (NH4 )2 SO4 Fe2 (SO4 )3 24H2 O, ч.д.а.
10. Аммоний сернокислый (NH4 )2 SO4 , ч.д.а.
11. Натрий уксуснокислый CH3 COONa, ч.д.а.
12. Алюминон (аммонийная соль ауринтрикарбоновой кислоты), ч.д.а.
13. Метаванадиевокислый аммоний NH4 VO3 , ч.д.а.
14. Вольфрамовокислый натрий 2-водный Na2 WO4 2H2 O, ч.д.а.
15. Титан треххлористый TiCl3 , ч.д.а.
16. Родамин В (тетраэтилдиамино-о-карбоксилфенилксантенилхлорид), ч.д.а.
17. Толуол C7 H8 , ч.д.а.
18. Бутилацетат C6 H12 O2 , ч.д.а.
2.5. Методики анализов
2.5.1. Фотометрическое определение железа с сульфосалициловой кислотой [39]
Метод основан на том, что сульфосалициловая кислота или её натриевая соль образует с солями железа окрашенные комплексные соединения. Причем в слабокислой среде сульфосалициловая кислота реагирует только с солями Fe (III) (красное окрашивание), а в слабощелочной - с солями Fe (III) и (II) (желтое окрашивание).
Реактивы:
1. Сульфосалициловая кислота, 10%-ный раствор, или сульфосалицилат натрия, насыщенный раствор.
2. Аммиак, разбавленный раствор. Смешивают 200 мл концентрированного аммиака и 300 мл дистиллированной воды.
3. Стандартный раствор соли железа. Растворяют 0,8634 г железоаммонийных квасцов в дистиллированной воде, к раствору добавляют 10 мл H2 SO4 плотностью 1,84 г/см3 , разбавляют в мерной колбе на 1 л. Отбирают 100 мл полученного раствора, разбавляют водой в мерной колбе снова до 1 л.
В 1 мл раствора содержится 0,01 мг железа.
Ход определения общего содержания железа.
В коническую колбу вместимостью 50 мл наливают 10 мл анализируемой воды. В этом объеме должно содержаться от 1 до 10 мкг железа, что соответствует концентрациям от 0,1 до 1 мг/л. Более концентрированные по содержанию железа сточные воды предварительно разбавляют в мерной колбе так, чтобы содержание железа в 10 мл полученного раствора было в указанных пределах. Затем в пробирку приливают 5 мл раствора сульфосалициловой кислоты и 5 мл раствора аммиака.
Измеряют оптическую плотность полученного раствора на длине волны 420-430 нм по отношению к холостому раствору. Молярный коэффициент поглощения равен 5,5103 .
Содержание железа находят по градуировочному графику, для построения которого наливают из микробюретки 1-10 мл стандартного раствора, разбавляют до 10 мл дистиллированной водой и продолжают как при анализе пробы.
Построение градуировочного графика.
Были приготовлены реактивы как указано в методике. Взяли 2,0; 4,0; 6,0; 8,0; 10,0 мл стандартного раствора. Фотометрировали на ФЭКе на длине волны 440 нм в кюветах 3 см. Данные приведены в таблице 2.5.1.
Таблица 2.5.1.
Зависимость оптической плотности растворов от концентрации железа
| С, мкг/мл | 0,4 | 0,8 | 1,2 | 1,6 | 2,0 |
| D1 | 0,075 | 0,17 | 0,26 | 0,36 | 0,44 |
| D2 | 0,075 | 0,16 | 0,25 | 0,36 | 0,44 |
| D3 | 0,080 | 0,17 | 0,26 | 0,35 | 0,44 |
Статистическая обработка проводилась на ЭВМ по методу наименьших квадратов. Получено уравнение регрессии:
y=2,310-1 x
rт =0,878, rр =1,000
b=2,310-1 ±1,310-2 .
Рисунок 2.5.1.
|
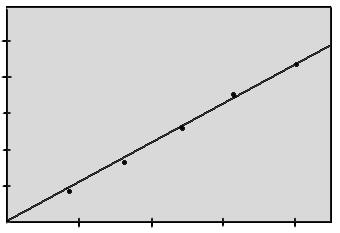 |
Градуировочный график поглощения растворов железа
|
2.5.2. Фотометрическое определение алюминия с алюминоном [40]
Метод основан на способности иона Al3+ образовывать с алюминием красное труднорастворимое комплексное соединение.
Реакция осуществляется при pH=4,9 в присутствии сульфата аммония. Оптическую плотность измеряют на длине волны 540 нм. Чувствительность метода 0,05 мг/л алюминия.
1. Приготовление основного стандартного раствора с концентрацией 0,10 г/мл. Стандартный раствор можно приготовить из алюминиевой стружки. Для этого 0,1 г стружки растворяют в 5 мл концентрированной соляной кислоты при нагревании и перемешивании. Переливают раствор в мерную колбу на 1000 мл и доводят до метки дистиллированной водой.
2. Приготовление рабочего стандартного раствора с концентрацией 0,005 мг/мл. 5,0 мл основного стандартного раствора помещают в мерную колбу на 100 мл и доводят объем до метки дистиллированной водой. Раствор готовят в день проведения анализа.
3. Приготовление раствора сульфата аммония. 50,0 г сульфата аммония растворяют в 100 мл дистиллированной воды.
4. Приготовление ацетатного буфера ( pH =4,9 ± 1). 241 г CH3 COONa помещают в колбу на 1000 мл и растворяют в 500 мл дистиллированной воды. Дают раствору остыть, добавляют 155 мл ледяной уксусной кислоты, доводят объем до метки дистиллированной водой. pH буфера устанавливают потенциометрически и, при необходимости, корректируют, добавляя NaOH или CH3 COOH. Для анализа исходный буферный раствор разбавляют в 10 раз дистиллированной водой. Срок хранения 3 месяца.
5. Приготовление раствора алюминона. 0,5 г алюминона растворяют в 125 мл горячей дистиллированной воды, затем добавляют 125 мл разбавленного ацетатного буфера. Раствор готов к употреблению сразу. Хранят его в склянке из темного стекла. Срок хранения 3 месяца.
6. Приготовление раствора гидроокиси натрия. 5,0 г NaOH растворяют в 100 мл дистиллированной воды.
7. Приготовление реакционной смеси. Смешивают одну объемную часть сульфата аммония с двумя частями алюминона и двадцатью двумя – разбавленного ацетатного буфера. В день анализа в необходимом объеме реакционной смеси растворяют аскорбиновую кислоту по 25 мг на каждые 25 мл реакционной смеси. Раствор можно хранить в течение 1 месяца.
Проведение анализа.
Проведению анализа может помешать окисное железо (Fe3+ ). Влияние железа массовой концентрации 0,3 мг/л и более устраняется восстановлением его аскорбиновой кислотой до закисного железа (Fe2+ ).
В мерную колбу на 50 мл, где уже находится исследуемый раствор, добавляют 25 мл реакционной смеси, 25-30 мг аскорбиновой кислоты, доводят до метки дистиллированной водой. Раствор выдерживают в течении 25-30 мин. Затем измеряют его оптическую плотность на длине волны 540 нм в кювете с толщиной слоя 30 мм. Измерения проводят относительно холостой пробы. Для приготовления холостой пробы в мерную колбу на 50 мл помещают 25 мл реакционной смеси, 25-30 мг аскорбиновой кислоты, доводят до метки дистиллированной водой. Количество алюминия в пробе находят по градуировочному графику. При необходимости производят разбавление исследуемого раствора.
Построение градуировочного графика.
Приготовление реактивов велось по методике. Взяли 0,5; 1,0; 3,0; 5,0; 7,0; 9,0 мл рабочего стандартного раствора. Фотометрировали на длине волны 540 нм в кюветах 3 см. Полученные данные приведены в таблице 2.5.2.
Таблица 2.5.2.
Зависимость оптической плотности растворов от концентрации алюминия
| С, мкг/мл | 0,05 | 0,1 | 0,3 | 0,5 | 0,7 | 0,9 |
| D1 | 0,04 | 0,08 | 0,22 | 0,36 | 0,50 | 0,66 |
| D2 | 0,03 | 0,07 | 0,20 | 0,34 | 0,48 | 0,64 |
| D3 | 0,04 | 0,07 | 0,21 | 0,34 | 0,48 | 0,64 |
Статистическая обработка проводилась на ЭВМ с помощью метода наименьших квадратов. Получено уравнение регрессии:
y=6,910-1 x
rт =0,878, rр =1,000
b=6,910-1 ±1,210-2 .
Рисунок 2.5.2.
Градуировочный график поглощения растворов алюминия
|
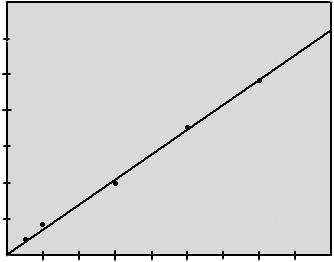 |
|
2.5.3. Фотометрическое определение ванадия [41]
1. Приготовление эталонного раствора ванадия 1000 мг/л.
В стакане вместимостью 100 мл растворяют 0,5740 г метаванадиевокислого аммония в 40 мл разбавленной азотной кислоты (1:4), количественно переносят в мерную колбу вместимостью 250 мл и доводят до метки дистиллированной водой.
2. Приготовление рабочего эталонного раствора ванадия 50 мг/л.
5 мл эталонного раствора ванадия 1000 мг/л количественно переносят в мерную колбу вместимостью 100 мл и доводят до метки дистиллированной водой.
3. Приготовление раствора вольфрамовокислого натрия 185 г/мл.
В стакане вместимостью 50 мл растворяют 18,5 г двуводного вольфрамовокислого натрия (Na2 WO4 2H2 O) в горячей дистиллированной воде, охлаждают до комнатной температуры, количественно переносят в мерную колбу вместимостью 100 мл и доводят до метки дистиллированной водой.
Проведение анализа.
В стакан вместимостью 100 мл помещают аликвоту раствора в зависимости от ожидаемого содержания ванадия, 2 мл разбавленной серной кислоты (1:1), 5 мл разбавленной ортофосфорной кислоты и 2,5 мл вольфрамовокислого натрия. Каждый раз содержимое стакана перемешивают встряхиванием. Раствор нагревают до 40-70°C, охлаждают до комнатной температуры, количественно переносят в мерную колбу вместимостью 50 мл, доводят до метки дистиллированной водой. Раствор должен быть прозрачным. Выдерживают в течение 60 мин. Затем измеряют его оптическую плотность на длине волны 440 нм в кювете с толщиной слоя 5 см. Измерения проводят относительно холостой пробы. Для приготовления холостой пробы в мерную колбу на 50 мл помещают 2 мл разбавленной серной кислоты (1:1), 5 мл разбавленной ортофосфорной кислоты и 2,5 мл вольфрамовокислого натрия, доводят до метки дистиллированной водой. Количество ванадия находят по градуировочному графику.
Построение градуировочного графика.
Приготовление растворов велось по методике. Взяли 1,0; 2,0; 4,0; 6,0; 8,0; 10,0 мл рабочего эталонного раствора. Фотометрировали на длине волны 440 нм в кюветах 5 см. Полученные данные приведены в таблице 2.5.3.
Таблица 2.5.3.
Зависимость оптической плотности растворов от концентрации ванадия
| С, мкг/мл | 1,0 | 2,0 | 4,0 | 6,0 | 8,0 | 10,0 |
| D1 | 0,06 | 0,12 | 0,26 | 0,39 | 0,50 | 0,62 |
| D2 | 0,05 | 0,12 | 0,26 | 0,38 | 0,50 | 0,62 |
| D3 | 0,05 | 0,11 | 0,25 | 0,38 | 0,50 | 0,60 |
Статистическая обработка проводилась на ЭВМ с помощью метода наименьших квадратов. Получено уравнение:
y=6,310-2 x
rт =0,811, rр =0,999
b=6,310-2 ±3,110-3 .
Рисунок 2.5.3.
|
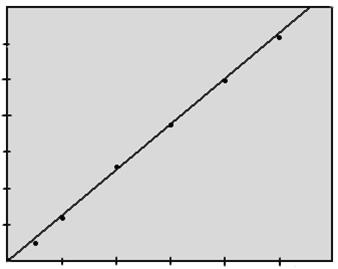 |
Градуировочный график поглощения растворов ванадия
|
2.5.4. Фотометрическое определение галлия [42]
Метод основан на образовании ионного ассоциата хлоргаллата родамина B и измерении оптической плотности его экстрактов. Интенсивная красно-фиолетовая окраска экстракта обеспечивает чувствительный способ определения малых количеств галлия. В этих условиях аналогично реагируют Sb (V), Au (III), Tl (III) и Fe (III). Вольфрам (VI) образует вольфрамат родамина B, не растворимый в толуоле.
Al, Zn, Cu, Ta и In не дают окраски при экстракции толуолом. Окислители, в том числе нитраты, должны отсутствовать в растворе. Сурьма и другие перечисленные металлы не реагируют с родамином B, если они восстановлены до более низких степеней валентности [Sb(III), Au(0), Tl(I), Fe(II)].
Реактивы:
1. Кислота соляная, 6-н. раствор.
2. Титан треххлористый, раствор, разбавленный концентрированной соляной кислотой 1:1.
3. Родамин B, 0,5%-ный раствор в 6-н. растворе соляной кислоты.
4. Смесь толуола с бутилацетатом. Готовят смешением толуола с бутилацетатом в соотношении 2:1.
5. Галлий металлический.
Приготовление стандартных растворов галлия.
1. Раствор А. Навеску галлия массой 0,1 г растворяют в 10 мл 6-н. раствора соляной кислоты при нагревании с добавлением нескольких капель пергидроля. Полученный раствор выпаривают досуха. Остаток снова растворяют в 6-н. растворе соляной кислоты, раствор переносят в мерную колбу вместимостью 100 мл, доливают 6-н. раствором соляной кислоты до метки и перемешивают. 1 мл раствора А содержит 1000 мкг галлия.
2. Раствор Б. Отбирают пипеткой 10 мл раствора А и переносят в мерную колбу вместимостью 100 мл, доливают 6-н. раствором соляной кислоты до метки и перемешивают. 1 мл раствора Б содержит 100 мкг галлия.
3. Раствор В. Отбирают пипеткой 10 мл раствора Б и переносят в мерную колбу вместимостью 100 мл, доливают 6-н. раствором соляной кислоты до метки и перемешивают. 1 мл раствора В содержит 10 мкг галлия.
4. Раствор Г. Отбирают пипеткой 10 мл раствора В и переносят в мерную колбу вместимостью 100 мл, доливают 6-н. раствором соляной кислоты до метки и перемешивают. 1 мл раствора Г содержит 1 мкг галлия.
Проведение анализа.
В мерную колбу вместимостью 50 мл (или делительную воронку вместимостью 25 мл) отбирают 5 мл анализируемого раствора и прибавляют по каплям при помешивании 1 мл треххлористого титана. Через 5 мин в колбу (или воронку) прибавляют 0,4 мл 0,5%-ного раствора родамина B, перемешивают и выдерживают в течение 5 мин. Затем приливают 5 мл смеси толуола с бутилацетатом, закрывают колбу (или воронку) притертой пробкой и встряхивают 2 мин. После разделения слоев осторожно сливают в кювету верхний органический слой, окрашенный в красно-фиолетовый цвет, и закрывают кювету крышкой. Колориметрирование производят на длине волны 540 нм в кювете с толщиной слоя 5 мм.
Нулевой раствор готовят следующим способом: в мерную колбу вместимостью 50 мл (или делительную воронку вместимостью 50 мл) наливают 10 мл 6-н. раствора соляной кислоты, приливают 2 мл треххлористого титана, 0,8 мл 0,5%-ного раствора родамина B, 10 мл смеси толуола с бутилацетатом, закрывают притертой пробкой колбу (или воронку) и встряхивают в течение 2 мин. После разделения органический слой сливают в кюветы для колориметрирования.
Построение градуировочного графика.
Приготовление реактивов велось по методике. Взяли 1,0; 2,0; 3,0; 4,0; 5,0 мл стандартного раствора Г. Фотометрировали на длине волны 540 нм в кюветах 5 мм. Данные приведены в таблице 2.5.4.
Таблица 2.5.4.
Зависимость оптической плотности растворов от концентрации галлия
| M, мкг | 2 | 4 | 6 | 8 | 10 |
| D1 | 0,09 | 0,13 | 0,19 | 0,26 | 0,27 |
| D2 | 0,10 | 0,14 | 0,18 | 0,26 | 0,30 |
| D3 | 0,11 | 0,14 | 0,18 | 0,23 | 0,29 |
Статистическая обработка проводилась на ЭВМ с помощью метода наименьших квадратов. Получено уравнение регрессии:
y=3,710-2 +2,510-2 x
rт =0,878, rр =0,983
a=3,710-2 ±5,710-2
b=2,510-2 ±8,610-3
Рисунок 2.5.4.
|
|
2.5.5. Методика определения кремния [43].
При растворении плава в соляной кислоте выделяется кремниевая кислота:
Na2 SiO3 +2HCl®2NaCl+H2 SiO3
Некоторая часть образовавшейся кремниевой кислоты остается в растворе в виде гидрозоля. Чтобы перевести ее полностью в осадок, раствор плава выпаривают и оcтаток от выпаривания высушивают, при этом золь кремниевой кислоты переходит в гель. При прокаливании до 1000°C кремнекислота теряет воду, превращаясь в кремниевый ангидрид:
H2
SiO3
![]() SiO2
+H2
O
SiO2
+H2
O
Выполнение определения.
Раствор в фарфоровой чашке после выщелачивания плава разбавляют 10%-ным раствором соляной кислоты до объема 150 мл и выпаривают досуха на водяной бане (избегать разбрызгивания). Затем добавляют 10 мл соляной кислоты (пл. 1,19) и снова выпаривают на водяной бане досуха. После этого сухой остаток сушат 1 ч в сушильном шкафу при температуре 120°C. Более эффективна сушка на водяной бане до полного удаления запаха HCl с последующим прокаливанием в течение 2 ч.
Содержимое чашки охлаждают и осторожно смачивают 15 мл HCl (пл. 1,19), прибавляя ее по каплям. Покрывают чашку часовым стеклом и оставляют стоять 10 мин. После этого содержимое чашки обрабатывают 70-80 мл горячей дистиллированной воды, дают стоять 10 мин на кипящей водяной бане и отстоявшийся раствор фильтруют через фильтр с красной лентой, не перенося осадка на фильтр. Осадок в чашке промывают горячей водой, вначале декантацией, затем переносят его на фильтр и промывают его на фильтре до исчезновения реакции на хлор с азотнокислым серебром в промывных водах.
Для удаления невидимых на белом фоне чашки оставшихся крупинок кремнекислоты чашку протирают маленьким кусочком фильтровальной бумаги, который присоединяют к осадку (№1) на фильтре. Фильтрат количественно переносят в ту же чашку, в которой проводилось выпаривание, и вторично выпаривают досуха и до удаления запаха HCl. После охлаждения сухой остаток в чашке смачивают по каплям соляной кислотой (пл. 1,19) в количестве 5-7 мл, накрывают часовым стеклом и оставляют стоять 10 мин. Затем содержимое чашки обрабатывают 30 мл горячей дистиллированной воды и дают стоять 10 мин на кипящей водяной бане и далее поступают так же, как и в первый раз: отфильтровывают, промывают и т. д.
Вторичной обработкой фильтрата достигают более полного перевода золя кремнекислоты в осадок. Фильтрат №1 сохраняют для дальнейшего анализа, а осадок №2 на фильтре присоединяют вместе с фильтром к ранее выделенному осадку №1 кремнекислоты.
Общий осадок кремнекислоты вместе с фильтрами озоляют вначале при низкой температуре во взвешенном и прокаленном платиновом или фарфоровом тигле на пламени газовой горелки, помещая тигель на асбестовую сетку, затем на голом огне, постепенно увеличивая пламя. Под конец прокаливают до постоянной массы в муфельной печи (в течение 30-50 мин при t=1000-1100°C). Весовая форма SiO2 белого цвета.
2.5.6. Выполнение сплавления и растворения навески силиката
(золы) [43]
Для сплавления навеску испытуемого материала (золы) в количестве 0,5-1,0 г отвешивают в фарфоровом тигле с точностью до 0,0002 г. К навеске добавляют 4-6 г безводной соды, хорошо растертой в фарфоровой ступке. Величина фарфорового тигля должна быть такой, чтобы он не более чем наполовину был заполнен порошком. Содержимое тигля осторожно и тщательно перемешивают маленьким шпателем, затем смесь сверху засыпают 1-2 г чистой соды.
После этого тигель вносят в разогретую до 200-300°C муфельную печь и медленно нагревают до 800-900°C в течение 30-40 мин. Затем при данной температуре выдерживают 1,5 ч. После охлаждения тигля плав переносят в фарфоровую чашку емкостью 200-250 мл и растворяют в 30-40 мл HCl (1:1). Тигель несколько раз ополаскивают раствором HCl (1:1), сливая раствор в фарфоровую чашку. Раствор в фарфоровой чашке после выщелачивания сплава выпаривают досуха, избегая разбрызгивания. Затем добавляют 10 мл соляной кислоты (пл. 1,19) и снова выпаривают досуха.
Содержимое чашки охлаждают и осторожно смачивают небольшим количеством HCl (1:1). Оставляют стоять 10 мин, нагревают и отстоявшийся раствор фильтруют, не перенося осадка на фильтр. Осадок в чашке промывают раствором HCl (1:1), вначале декантацией, затем переносят его на фильтр и промывают на фильтре. Полученный раствор анализируют.
2.6. Методики эксперимента
2.6.1. Методика обескремнивания
В стеклянный термостойкий стакан вместимостью 1000 мл помещается 800 мл щелочного раствора концентрацией 200 г/л. Раствор термостатируется до 80°C. Затем в стакан помещается 160 г золы и включается механическая мешалка. Время обескремнивания 3 часа.
2.6.2. Методика щелочной и кислотной обработки
Обработка золы проводится в термостойком стакане вместимостью 250 мл. Предварительно раствор едкого натра или серной кислоты термостатируется до необходимой температуры. Затем помещается зола и включается механическая мешалка.
2.6.3. Методика электрохимического выщелачивания
|
Для исследования была собрана электрохимическая ячейка:
В электрохимическую ячейку помещается серная кислота или раствор едкого натра объемом 200-250 мл и концентрацией 100-400 г/л и, при необходимости, термостатируется. После этого в ячейку помещается навеска (20-25 г) обескремненной золы, включается механическая мешалка и электролизер. Сила тока задается в пределах 1-10 А.
3. ПОЛУЧЕННЫЕ РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
3.1. Отработка методики анализа на содержание ванадия
Ванадий определялся по методике ГОСТ 10364-90. Она предусмотрена для определения ванадия (V).Но в наших опытах при электролизе ванадийсодержащих растворов ванадий может перейти в более низкую степень окисления (III, IV). Поэтому нами предварительно отрабатывалась методика окисления ванадия.
Опыты проводились с модельными растворами NH4 VO3 , которые готовились следующим образом. Навеска NH4 VO3 массой 0,5740 г растворялась в растворе H2 SO4 (1:1), и определялось содержание ванадия по методике. Оно составило 50,9±5,8 мг/л (статистическая обработка в приложении 1).
Затем этот раствор восстанавливали цинком при кипячении в течение 0,5 ч до V(II) фиолетовой окраски. Анализ показал содержание ванадия 32,5±1,6 мг/л, что дает заниженные результаты.
Для окисления V(II) до V(V) использовалась концентрированная азотная кислота в расчете 5 мл HNO3 на 100 мл раствора. Анализ окисленного раствора по методике показал значение 49,9±2,3 мг/л.
Таким образом, предварительное окисление аликвоты азотной кислотой позволяет определить истинное содержание ванадия в растворе.
3.2. Поведение галлия и ванадия при выщелачивании
Предварительными исследованиями было показано, что обработка золы ТЭЦ раствором щелочи 200 г/л при температуре 80°C и времени выщелачивания 2 ч приводит к извлечению 49,7% кремния, 5,5% алюминия и 5,6% ванадия. То есть такая обработка приводит к концентрированию металлов в зольном остатке. Удаление аморфной части SiO2 , частичное разрушение частиц золы должно обеспечить более эффективное извлечение галлия и ванадия из золы. Таким образом обработанная зола использовалась в последующих опытах.
3.2.1. Исследование влияния температуры и добавки NaCl на эффективность выщелачивания галлия, ванадия, железа и алюминия в сернокислой среде
Исследование проводилось при условиях:
· концентрация H2 SO4 200 г/л;
· время обработки 2 ч;
· Т:Ж=1:5 (40 г золы и 200 мл серной кислоты).
Анализ на содержание элементов в растворе проводился по методикам 2.5.1 – 2.5.4. Степень извлечения металлов рассчитывалась исходя из их содержания в спеке обескремненной золы, %: Ga – 0,0036; V – 0,033; Fe – 5,77; Al – 17,9 (см. 2.5.6.). Данные приведены в таблице 3.2.1.
Таблица 3.2.1.
Полнота выщелачивания Ga, V, Fe и Al в серной кислоте
| Темпе-ратура, °C | NaCl, г/л | Ga | V | Fe | Al | ||||
| m, мг | a, % | m, мг | a, % | m, г | a, % | m, г | a, % | ||
| 55 | - | 0,09±0,02 | 6,1 | 0,81±0,11 | 4,6 | 0,66±0,02 | 36,8 | 0,78±0,05 | 17,7 |
| 70 | - | 0,14±0,04 | 9,6 | 1,09±0,15 | 6,2 | 0,85±0,03 | 47,2 | 0,89±0,06 | 20,2 |
| 90 | - | 0,23±0,06 | 16,3 | 1,06±0,15 | 6,0 | 1,09±0,04 | 60,4 | 1,04±0,06 | 23,6 |
| 85 | 5 | 0,37±0,10 | 25,6 | 1,02±0,14 | 5,8 | 1,28±0,05 | 71,1 | 1,03±0,06 | 23,4 |
| 85 | 50 | 0,35±0,10 | 24,0 | 1,23±0,17 | 7,0 | 1,44±0,05 | 79,8 | 0,92±0,06 | 20,8 |
Статистическая обработка результатов в приложении 2.
Из таблицы 3.2.1. видно, что кислотное выщелачивание не приводит к эффективному извлечению галлия и ванадия из золы-уноса ТЭЦ. Повышение температуры и введение добавки NaCl увеличивает извлечение галлия в 2,7 и 4 раза соответственно. Но полнота извлечения галлия недостаточна. Можно отметить значительную степень извлечения железа серной кислотой, которая при температуре 80°C и в присутствии NaCl концентрацией 50 г/л достигает 79,8%, что может быть использовано при разработке технологической схемы комплексной переработки золы.
3.2.2. Исследование эффективности выщелачивания галлия, ванадия, железа и алюминия щелочными растворами с добавкой NaCl
Обработка обескремненной золы в щелочной среде проводилась в условиях:
· концентрация щелочи 200 г/л;
· температура 80°C;
· время обработки 2 ч;
· Т:Ж=1:5 (40 г золы и 200 мл щелочи).
Полученные данные приведены в таблице 3.2.2.
Таблица 3.2.2.
Извлечение Ga, V, Fe и Al при щелочной обработке
| NaCl, г/л | Ga | V | Fe | Al | ||||
| m, мг | a, % | m, мг | a, % | m, мг | a, % | m, мг | a, % | |
| - | 0,81±0,09 | 56,3 | 2,1±0,4 | 12,1 | 23,4±1,0 | 1,3 | 339±11 | 7,7 |
| 5 | 0,79±0,09 | 55,1 | 1,2±0,2 | 7,0 | 12,4±0,5 | 0,7 | 66±2 | 1,5 |
| 50 | 0,80±0,09 | 56,0 | 1,8±0,3 | 10,2 | 7,9±0,3 | 0,4 | 53±2 | 1,2 |
Статистическая обработка результатов в приложении 3.
Из таблицы 3.2.2. следует, что при обработке щелочью железо и алюминий выщелачиваются незначительно; ванадий выщелачивается на 12%, что в 2 раза больше, чем в кислоте, но также недостаточно. Довольно эффективно в щелочной среде переходит в раствор из золы галлий, более чем наполовину. Введение добавки NaCl уменьшает степень извлечения ванадия и алюминия и практически не сказывается на степени извлечения галлия и железа.
3.2.3. Исследование влияния концентрации щелочи, температуры и времени выщелачивания на полноту извлечения галлия
В связи с тем, что в щелочной среде галлий выщелачивается эффективно, было изучено влияние на степень извлечения галлия из необработанной золы таких факторов, как концентрация щелочи, температура и время выщелачивания (таблица 3.2.3.1.). Было проведено математическое планирование эксперимента и составлен план 23 (таблица 3.2.3.2.). Степень извлечения галлия рассчитывалась исходя из его содержания в спеке необескремненной золы: 0,0035%.
Таблица 3.2.3.1.
Факторное пространство
| № | Фактор | Центр плана | Интервал варьирования | Погрешность |
| I | Температура (X1 ), °C | 70 | 20 | 2 |
| II | Концентрация щелочи (X2 ), г/л | 250 | 150 | 5 |
| III | Время выщелачивания (X3 ), ч | 2,5 | 1,5 | 0,1 |
Таблица 3.2.3.2.
План эксперимента и полученные результаты
| № | Температура (X1 ), °C | Концентрация щелочи (X2 ), г/л | Время выщелачивания (X3 ), ч | mGa , мкг | aGa , % |
| 1 | 90 (+) | 400 (+) | 4 (+) | 306±32 | 47,8 |
| 2 | 50 (-) | 400 (+) | 4 (+) | 146±17 | 22,8 |
| 3 | 90 (+) | 100 (-) | 4 (+) | 36,3±6,6 | 5,6 |
| 4 | 50 (-) | 100 (-) | 4 (+) | 15,1±2,8 | 2,4 |
| 5 | 90 (+) | 400 (+) | 1 (-) | 214±24 | 33,4 |
| 6 | 50 (-) | 400 (+) | 1 (-) | 76,4±10,1 | 11,8 |
| 7 | 90 (+) | 100 (-) | 1 (-) | 24,1±2,8 | 3,8 |
| 8 | 50 (-) | 100 (-) | 1 (-) | 11,2±1,2 | 1,8 |
Статистическая обработка результатов в приложении 4.
Получено уравнение регрессии:
aGa , %=16,2+6,5X1 +12,8X2 +3,5X3 +5,2X1 X2 +2,9X2 X3
Таким образом, из полученных коэффициентов уравнения регрессии можно сделать следующие выводы. Наибольшее влияние на степень извлечения галлия оказывает концентрация щелочи, меньшее – температура и время выщелачивания. При этом совместное влияние температуры и концентрации щелочи значительнее, чем сочетание концентрации и времени выщелачивания.
Для изучения влияния времени на насыщаемость раствора галлием при обработке обескремненной золы щелочным раствором нами были выбраны следующие условия:
· концентрация щелочи 200 г/л;
· температура 80°C;
· Т:Ж=1:5 (20 г золы и 100 мл щелочи).
Полученные результаты приведены в таблице 3.2.3.3.
Таблица 3.2.3.3.
Зависимость степени извлечения галлия от времени
Время выщелачивания, ч |
Количество извлеченного галлия, мг | Степень извлечения a, % |
| 1 | 0,35±0,06 | 48,6 |
| 2 | 0,41±0,05 | 56,3 |
| 3 | 0,43±0,09 | 60,2 |
| 4 | 0,48±0,06 | 66,9 |
Статистическая обработка результатов в приложении 5.
Рисунок 3.2.3.
|
|
Из таблицы 3.2.3.3 и рисунка 3.2.3. видно, что зависимость прямолинейная и максимум извлечения за 4 часа не достигнут. При этом за первый час в раствор переходит почти половина галлия.
3.2.4. Исследование возможности полного извлечения галлия при многостадийной обработке золы
Для того чтобы интенсифицировать процесс выщелачивания галлия, обескремненная зола последовательно обрабатывалась свежими растворами щелочи в 3 стадии по одному часу.
Исследование проводилось в условиях:
· концентрация щелочи 200 г/л;
· температура 80°C;
· Т:Ж=1:5 (20 г золы и 100 мл щелочи).
Данные приведены в таблице 3.2.4.
Таблица 3.2.4.
Полнота извлечения галлия при многократной обработке растворами щелочи
| Стадия | Время обработки, ч | S количество извлеченного галлия, мг | SaGa , % |
| I (обескремнивание) | 3 | 1,25±0,14 | 63,5 |
| II | 1 | 1,61±0,17 | 81,7 |
| III | 1 | 1,85±0,18 | 94,1 |
| IV | 1 | 1,93±0,21 | 98,2 |
Статистическая обработка результатов в приложении 6.
Таким образом, при многократной обработке обескремненной золы галлий извлекается практически полностью.
3.3. Электрохимическое выщелачивание ванадия
Поскольку в предыдущих опытах не была достигнута достаточная степень извлечения ванадия в раствор, нами исследовалось электрохимическое выщелачивание в кислой и щелочной средах (см. 2.6.3.).
3.3.1. Электровыщелачивание в кислой среде
Исследования проводились в условиях:
· концентрация H2 SO4 100 г/л;
· температура 20°C;
· время электролиза 2 ч;
· отношение Т:Ж=1:10 (20 г золы и 200 мл кислоты);
· титановый катод и свинцовый анод.
Полученные данные приведены в таблице 3.3.1.
Таблица 3.3.1.
Зависимость полноты извлечения металлов от плотности тока
| Условия опыта | V | Fe | Al | |||
| m, мг | a, % | m, г | a, % | m, г | a, % | |
Sk =41 см2 I=1 A i=24,4 mA/см2 |
0,26±0,04 | 3,9 | 0,30±0,02 | 25,7 | 0,32±0,04 | 8,9 |
I=2 A i=48,8 mA/см2 |
0,38±0,06 | 5,8 | 0,33±0,03 | 29,0 | 0,37±0,04 | 10,2 |
I=5 A i=122 mA/см2 |
0,73±0,11 | 11,0 | 0,39±0,03 | 33,8 | 0,40±0,05 | 11,3 |
I=10 A i=244 mA/см2 |
0,77±0,11 | 11,7 | 0,59±0,05 | 51,5 | 0,45±0,05 | 12,7 |
Статистическая обработка результатов в приложении 7.
Из таблицы 3.3.1. следует, что повышение плотности тока с 24,4 до 244 mA/см2 увеличивает степень извлечения железа на 25%, алюминия – на 4%, ванадия – на 8%, что все же недостаточно.
3.3.2. Электровыщелачивание в щелочной среде
Было исследовано влияние температуры на электровыщелачивание ванадия в щелочной раствор. Исследование проводилось при условиях:
· концентрация щелочи 200 г/л;
· время обработки 1 ч;
· отношение Т:Ж=1:10 (25 г золы и 250 мл щелочи);
· титановые катод и анод;
· i=25 mA/см2 .
Полученные данные приведены в таблице 3.3.2.
Таблица 3.3.2.
Зависимость содержания ванадия в растворе электровыщелачивания от температуры
| t, °C | mV , мг | aV , % |
| 20 | 0,67±0,15 | 8,1 |
| 85 | 1,05±0,23 | 12,7 |
Из анализа таблиц 3.3.1. и 3.3.2. можно заключить, что как в кислой, так и в щелочной средах в исследованных условиях не наблюдается увеличения степени извлечения ванадия. Поэтому поиски по извлечению ванадия должны быть продолжены.
ВЫВОДЫ
1. Показано, что галлий может быть полностью извлечен из золы-уноса ТЭЦ четырехкратной обработкой раствором щелочи концентрацией 200 г/л при t=80°C и Т:Ж=1:10.
2. Извлечение ванадия из золы-уноса ТЭЦ растворами кислоты и щелочи в исследованных условиях составляет 11-12%.
3. Добавка NaCl при кислотном выщелачивании позволяет извлечь в раствор 79,8% железа и 24% галлия.
ЛИТЕРАТУРА
1. Дымов А.М., Савостин А.П. Аналитическая химия галлия. – М.: Наука, 1968. – С. 5-9, 131-134.
2. Иванова Р.В. Химия и технология галлия. – М.: Металлургия, 1973. – С. 5-9, 131-134.
3. Еремин Н.И. Галлий. – М.: Металлургия, 1964.
4. Меерсон Г.А., Зеликман А.Н. Металлургия редких металлов. – М.: Металлургииздат, 1955. – С. 495-501.
5. Beja M. Chimie et Industrie, 67, I, 1952, 45-55, цит. по [4].
6. Резник П.А., Иванова Р.В. Сборник научных трудов Гиредмета, 1, 238, 258 (1959).
7. Резник П.А., Миронова З.М. Цветные металлы, 12, 60 (1940).
8. Яценко С.П., Деменев Н.В. Журнал неорганической химии, 4, 869 (1959).
9. Хасиева С.А., Зеликман А.Н., Иванова Р.В. Азербайджанский химический журнал, №5, 109 (1964).
10. Еремин Н.И., Гуськов В.М. Журнал прикладной химии, 33, 157 (1960).
11. Нижник А.Т., Шехтер З.В. Журнал прикладной химии, 35, 295 (1962).
12. Шалавина Е.Л., Гусарова Т.Д. Труды института металлургии АН КазССР, 1964. – Т. 9. – С. 121-129.
13. Шалавина Е.Л., Гусарова Т.Д. Труды института металлургии АН КазССР, 1965. – Т. 12. – С. 52-57.
14. Химия и технология редких и рассеяных элементов. Ч. I. /Под ред. К.А. Большакова. – М.: Высшая школа, 1976. – С. 245-276.
15. Gastinger E. Berg-und Huttenmannische Monatshefte, 99, I, 1954, 13, цит. по [4].
16. Нижник А.Т., Шехтер З.В. Журнал прикладной химии, 37, 742 (1964).
17. Morgan G.J. Chem. Soc., 1935, p.556, цит. по [2].
18. Кострикин В.М., Иванов-Эмин Б.Н. Журнал прикладной химии, 13, №10 (1940).
19. Janagari M.J. Coal Research Inst. (Japan), 1956, v. 7, p. 129-138, цит. по [2].
20. Schreiter W. Chem. Techn., 1954, Bd 3, s. 141-143, цит. по [2].
21. Еремин Н.И. Изв. вузов. Цветная металлургия. 1960. Т. 2. С. 108.
22. Абишева З.С., Блайда И.А., Пономарева Е.И. Цветные металлы. 1994. №3. С. 36-38.
23. Абишева З.С., Блайда И.А., Пономарева Е.И. Цветные металлы. 1994. №2. С. 42-44.
24. Людоговский Г.И. Требования промышленности к качеству минерального сырья. Ванадий. Госгеолтехиздат, 1960.
25. Химия и технология редких и рассеянных элементов. Ч. III. /Под ред. К.А. Большакова. – М.: Высшая школа, 1976. – С. 16-36.
26. Ежовска-Тршебятовска Б., Копач С., Микульский Т. Редкие элементы. – М.: Мир, 1979. – С. 297-300.
27. Амирова С.А., Печковский В.В. и др. Журнал прикладной химии, 36, №5, 936 (1963).
28. Veres J. Acta techn. Acad. scient. hung., 41, №3-4 (1962), цит. по [25].
29. Винаров И.В., Янкелевич Р.Г. Украинский химический журнал, 30, №5, 524 (1964).
30. Кунаев А.М. Пиро-гидрометаллургические способы переработки ванадиевого сырья Казахстана. – Алма-Ата: Наука, 1971. – С. 17-21.
31. Основы металлургии. Т. IV. Редкие металлы. /Отв. ред. Грейвер Н.С. и др. – М.: Металлургиздат, 1967.
32. Ростокер У. Металлургия ванадия. – М.: ИЛ., 1959. – С. 9.
33. Izumi Tsuboi, Shigetami Kasai, Takuya Yamamoto, Isao Komasawa, Eiichi Kunugita. Извлечение редких металлов из летучей угольной золы // Int. Solv. Extr. Conf., 1990 (ISEC’90), Kyoto, July 16-21, 1990: Abstr.–[Kyoto], 1990. – С. 215. – Англ., цит. по РЖХим 8Л52, 1991.
34. Schemel Roberto, Rodriguez Domingo, Salazar Ramon. Выщелачивание и извлечение ванадия из ванадийсодержащих отходов //Пат. 4539186, США. Заявл. 15.03.84 №589951, опубл. 3.09.85. МКИ С 01 G 31/00, НКИ 423/62, цит. по РЖХим 12Л121, 1986.
35. Каваёси Яцухиро. Извлечение ванадия // Заявка 60-161339, Япония. Заявл. 30.01.84, №59-13332, опубл. 23.08.85. МКИ С 01 G 31/00, С 22 В 34/22, цит. по РЖХим 13Л126, 1986.
36. Патент №4798709, США, опубл. 17.01.89. МКИ С 01 G 31/00, НКИ 423-63. Способ обработки золы-уноса.
37. Поляков А.Ю. Основы металлургии ванадия. – М.: Металлургиздат, 1959.
38. Слотвинский-Сидак Н.Г., Потапов И.В. Изв. вузов. Цветная металлургия. 1962. №3. С.100-107.
39. Лурье Ю.Ю. Аналитическая химия промышленных сточных вод. – М.: Химия, 1984. – С. 111.
40. ГОСТ 18165-81. Вода питьевая. Метод определения массовой концентрации алюминия. – М., 1981.
41. ГОСТ 10364-90. Нефть и нефтепродукты. Метод определения ванадия. – М., 1990.
42. ГОСТ 12711-77. Твердое топливо. Метод определения массовой доли галлия. – М., 1977.
43. Годовская К.И., Рябина Л.В., Новиков Е.Ю., Гернер М.М. Технический анализ. – М.: Высшая школа, 1967. – С. 386-387.
ПРИЛОЖЕНИЯ
Приложение 1
Статистическая обработка результатов определения ванадия в модельном растворе
| Концентрация ванадия | C1 | C2 | C3 | C4 | C5 | C6 | |
| I | в исходном растворе, мг/л | 52,5 | 40,5 | 49,9 | 56,3 | 51,6 | 54,6 |
| II | после восстановления цинком, мг/л | 31,0 | 32,6 | 23,5 | 31,8 | 34,5 | 32,4 |
| III | в окисленном растворе, мг/л | 51,9 | 51,8 | 48,3 | 49,5 | 32,0 | 48,2 |
Статистические характеристики выборок:
P=0,95
| I | II | III | |
| Грубых промахов по Q-тесту не обнаружено | 23,5 – грубый промах, при QТ абл =0,640 дает Qэкс =0,682 | 32,0 – грубый промах, при QТабл =0,640 дает Qэкс =0,814 | |
| N | 6 | 5 | 5 |
| Xср | 50,9 | 32,5 | 49,9 |
| D | 31,01 | 1,69 | 3,30 |
| S | 5,57 | 1,30 | 1,82 |
| S |
2,27 | 0,58 | 0,81 |
| Sr | 0,11 | 0,04 | 0,04 |
| W, % | 10,9 | 4,0 | 3,6 |
| tст | 2,57 | 2,78 | 2,78 |
| Полуширина доверительного интервала | 5,8 | 1,6 | 2,3 |
| Границы доверительного интервала | 45,1 – 56,7 | 30,8 – 34,1 | 47,7 – 52,2 |
Приложение 2
Статистическая обработка результатов анализа на галлий, ванадий, железо, алюминий в растворе выщелачивания при температуре 55°C
| Количество извлеченного металла | m1 | m2 | m3 |
| Ga, мг | 0,09 | 0,10 | 0,08 |
| V, мг | 0,85 | 0,76 | 0,82 |
| Fe, г | 0,65 | 0,67 | 0,66 |
| Al, г | 0,80 | 0,76 | 0,79 |
Статистические характеристики выборок:
N=3
P=0,95
Грубых промахов по Q-тесту не обнаружено.
| Ga | V | Fe | Al | |
| Xср | 0,09 | 0,81 | 0,66 | 0,78 |
| D | 0,0001 | 0,002 | 0,0001 | 0,0004 |
| S | 0,01 | 0,05 | 0,01 | 0,02 |
| S |
0,006 | 0,03 | 0,006 | 0,01 |
| Sr | 0,111 | 0,057 | 0,015 | 0,027 |
| W, % | 11,1 | 5,7 | 1,5 | 2,7 |
| tст | 4,30 | 4,30 | 4,30 | 4,30 |
| полуширина доверительного интервала | 0,02 | 0,11 | 0,02 | 0,05 |
| границы доверительного интервала | 0,07 – 0,11 | 0,70 – 0,92 | 0,64 – 0,68 | 0,73 – 0,84 |
Приложение 3
Статистическая обработка результатов анализа на галлий, ванадий, железо, алюминий в растворе щелочной обработки
| Количество извлеченного металла | m1 | m2 | m3 |
| Ga, мг | 0,81 | 0,84 | 0,77 |
| V, мг | 2,1 | 1,9 | 2,2 |
| Fe, мг | 23,9 | 23,1 | 23,3 |
| Al, мг | 344 | 338 | 335 |
Статистические характеристики выборок:
N=3
P=0,95
Грубых промахов по Q-тесту не обнаружено.
| Ga | V | Fe | Al | |
| Xср | 0,81 | 2,1 | 23,4 | 339 |
| D | 0,001 | 0,02 | 0,17 | 21,0 |
| S | 0,04 | 0,15 | 0,42 | 4,58 |
| S |
0,02 | 0,09 | 0,24 | 2,65 |
| Sr | 0,04 | 0,07 | 0,02 | 0,01 |
| W, % | 4,35 | 7,39 | 1,78 | 1,35 |
| tст | 4,30 | 4,30 | 4,30 | 4,30 |
| полуширина доверительного интервала | 0,09 | 0,4 | 1,0 | 11 |
| границы доверительного интервала | 0,72 – 0,89 | 1,7 – 2,4 | 22,4 – 24,5 | 328 – 350 |
Приложение 4
Статистическая обработка результатов планирования
| № опыта | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| mGa , мкг | ||||||||
| m1 | 292 | 153 | 35,8 | 13,8 | 212 | 72,4 | 23,8 | 10,8 |
| m2 | 318 | 139 | 39,2 | 15,9 | 206 | 76,2 | 25,4 | 11,7 |
| m3 | 307 | 145 | 34,0 | 15,5 | 225 | 80,5 | 23,2 | 11,0 |
Статистические характеристики выборок:
N=3
P=0,95
Грубых промахов по Q-тесту не обнаружено.
| № опыта | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| Xср | 306 | 146 | 36,3 | 15,1 | 214 | 76,4 | 24,1 | 11,2 |
| D | 170 | 49,3 | 6,97 | 1,24 | 94,3 | 16,4 | 1,29 | 0,22 |
| S | 13,1 | 7,02 | 2,64 | 1,12 | 9,71 | 4,05 | 1,14 | 0,47 |
| S |
7,54 | 4,06 | 1,52 | 0,64 | 5,61 | 2,34 | 0,66 | 0,27 |
| Sr | 0,04 | 0,05 | 0,07 | 0,07 | 0,04 | 0,05 | 0,05 | 0,04 |
| W, % | 4,27 | 4,82 | 7,27 | 7,40 | 4,53 | 5,31 | 4,71 | 4,23 |
| tст | 4,30 | 4,30 | 4,30 | 4,30 | 4,30 | 4,30 | 4,30 | 4,30 |
| полуширина доверительного интервала | 32 | 17 | 6,6 | 2,8 | 24 | 10,1 | 2,8 | 1,2 |
| границы доверительного интервала | 273 – 338 | 128 – 163 | 29,8 – 42,9 | 12,3 – 17,8 | 190 – 238 | 66,3 – 86,4 | 21,3 – 26,9 | 10,0 – 12,3 |
Приложение 5
Статистическая обработка результатов анализа на галлий в растворах выщелачивания
| t, ч | 1 | 2 | 3 | 4 |
| mGa , мкг | ||||
| m1 | 0,35 | 0,40 | 0,43 | 0,45 |
| m2 | 0,38 | 0,43 | 0,47 | 0,50 |
| m3 | 0,33 | 0,39 | 0,40 | 0,48 |
Статистические характеристики выборок:
N=3
P=0,95
Грубых промахов по Q-тесту не обнаружено.
| t, ч | 1 | 2 | 3 | 4 |
| Xср | 0,35 | 0,41 | 0,43 | 0,48 |
| D | 0,0006 | 0,0004 | 0,001 | 0,0006 |
| S | 0,03 | 0,02 | 0,04 | 0,03 |
| S |
0,01 | 0,01 | 0,02 | 0,01 |
| Sr | 0,07 | 0,05 | 0,08 | 0,05 |
| W, % | 7,12 | 5,12 | 8,10 | 5,28 |
| tст | 4,30 | 4,30 | 4,30 | 4,30 |
| полуширина доверительного интервала | 0,06 | 0,05 | 0,09 | 0,06 |
| границы доверительного интервала | 0,29 – 0,41 | 0,36 – 0,46 | 0,35 – 0,52 | 0,41 – 0,54 |
Приложение 6
Статистическая обработка результатов анализа на галлий при многократной обработке растворами щелочи
| стадия | I | II | III | IV |
| mGa , мкг | ||||
| m1 | 1,27 | 1,63 | 1,84 | 1,95 |
| m2 | 1,19 | 1,54 | 1,93 | 2,01 |
| m3 | 1,30 | 1,67 | 1,79 | 1,84 |
Статистические характеристики выборок:
N=3
P=0,95
Грубых промахов по Q-тесту не обнаружено.
| Стадия | I | II | III | IV |
| Xср | 1,25 | 1,61 | 1,85 | 1,93 |
| D | 0,003 | 0,004 | 0,005 | 0,007 |
| S | 0,06 | 0,07 | 0,07 | 0,09 |
| S |
0,03 | 0,04 | 0,04 | 0,05 |
| Sr | 0,04 | 0,04 | 0,04 | 0,04 |
| W, % | 4,54 | 4,13 | 3,83 | 4,46 |
| tст | 4,30 | 4,30 | 4,30 | 4,30 |
| Полуширина доверительного интервала | 0,14 | 0,17 | 0,18 | 0,21 |
| границы доверительного интервала | 1,11 – 1,39 | 1,45 – 1,78 | 1,68 – 2,03 | 1,72 – 2,15 |
Приложение 7
Статистическая обработка результатов анализа на ванадий, железо и алюминий в растворах электровыщелачивания
| Количество извлеченного металла | m1 | m2 | m3 |
| V, мг | 0,26 | 0,24 | 0,27 |
| Fe, г | 0,30 | 0,29 | 0,31 |
| Al, г | 0,33 | 0,30 | 0,32 |
Статистические характеристики выборок:
N=3
P=0,95
Грубых промахов по Q-тесту не обнаружено.
| V | Fe | Al | |
| Xср | 0,26 | 0,30 | 0,32 |
| D | 0,0002 | 0,0001 | 0,0002 |
| S | 0,02 | 0,01 | 0,02 |
| S |
0,009 | 0,006 | 0,009 |
| Sr | 0,06 | 0,03 | 0,05 |
| W, % | 5,95 | 3,33 | 4,82 |
| tст | 4,30 | 4,30 | 4,30 |
| полуширина доверительного интервала | 0,04 | 0,02 | 0,04 |
| границы доверительного интервала | 0,22 – 0,29 | 0,28 – 0,32 | 0,28 – 0,35 |