Химия гидразина
СОДЕРЖАНИЕ: Химия и получение гидразина. Восстановление соединений, содержащих связь азот-азот. Получение из азотоводородной кислоты и азидов. Разложение аммиака. Синтез Рашига. Строение молекулы и дипольный момент. Монозамещенные и дизамещенные гидразины.Федеральное агентство по образованию
Государственное образовательное учреждение высшего профессионального
образования «Челябинский государственный университет»
Химический факультет
Химия гидразина.
Курсовая работа
Выполнила студентка
группы Х-302
Воробьева Ольга
Научный руководитель:
Белик В.А.
Работа защищена
« » __________________
Оценка_________________
Челябинск, 2007 год
ВВЕДЕНИЕ
Химия гидразина изучается уже почти три четверти века. До 1875 г. были известны только симметричные дизамещенные гидразина— гидразосоединения. В 1875 г. Э. Фишер, исследуя процесс восстановления диазосоединений, выделил и охарактеризовал простые органические производные гидразина. Он получил некоторые простые арилгидразины и охарактеризовал не только свободный фенилгидразин, но также и соли этого азотистого основания. Продукты восстановления азосоединений были названы гидразосоединениями; поэтому Фишер назвал типовое вещество N2Н4 гидразином и говорил о производных этого азотоводорода как о замещенных гидразинах. Область органических производных гидразина в дальнейшем также разрабатывалась Фишером, которому в течение последующих лет удалось синтезировать моно- и дизамещенные алкил- и арилгидразины. Исследуя свойства несимметричных дизамещенных гидразинов, Фишер показал, что они могут подвергаться окислению с образованием производных тетразена, одного из гипотетических цепочечных азотоводородов.
В 1887 г. Куртиусу и его сотрудникам удалось выделить гидразин и некоторые его соли. В связи с интенсивным развитием химии гидразина (азотистого аналога этана), которое имело место в течение последующих двадцати лет, Виланд в 1913 г. писал: «Систематическому развитию химии гидразина, изученной в настоящее время, достаточно полно, в значительной мере способствовало открытие вещества. Практически все комбинации гидразина с различными типами органических соединений могли быть затем получены из этого весьма реакционноспособного вещества. Производные гидразина вскоре превысили по числу и многообразию органические производные аммиака, которые изучались в течение многих десятилетий».
Гидразин является одним из простейших азотоводородов. Если сравнить этот класс соединений с углеводородами, то гидразин можно рассматривать как аналог этана. Он является вторым азотоводородом, выделенным в свободном состоянии. До настоящего времени в свободном состоянии было получено всего лишь три азотоводорода, а именно аммиак, гидразин и азид водорода. Другие азотоводороды давно известны в виде органических производных, и многие из них могут быть получены только в результате окисления соответствующих производных гидразина.
1.ПОЛУЧЕНИЕ ГИДРАЗИНА
Гидразин впервые был получен в виде органических производных. В 1887 г. Куртиус синтезировал и выделил неорганические соли, а также гидрат гидразина. Первые методы синтеза солей гидразина, из которых удалось получить его гидрат, были основаны главным образом на восстановлении соединений, содержащих связь азот—азот. Лишь позднее были предприняты попытки использовать в качестве исходного вещества аммиак и получать гидразин путем разложения или окисления аммиака и его производных.
Реакции, которые приводят к образованию гидразина, практического применения не получили. Синтез Рашига, включающий в себя частичное «окисление» аммиака (и мочевины) гипохлоритом, является единственным препаративным методом, который применяется для получения гидразина в производственных масштабах.
1.1 Восстановление соединений, содержащих связь азот-азот
Теоретически любое соединение, содержащее связь азот—азот, может быть восстановлено до гидразина или его производного. Соответствующие методы получили широкое применение в органической химии для получения органических производных гидразина; некоторые из них в результате последующей обработки дают соли гидразина или сам гидразин. Так, например, гидразин был получен из азотноватистой кислоты и ее изомеров, нитрамида и нитрозо-гидроксиламина, из бимолекулярных нитрозосоединений, а также из нитрозоаминов, азосоединений и азидов. Кроме того, в качестве исходных веществ были использованы нитриты, нитраты и другие нитросоединения, однако восстановление их, вероятно, протекает с образованием промежуточных соединений, содержащих связь азот—азот. Утверждали даже, что при некоторых условиях молекулярный азот может реагировать с водородом, образуя гидразин; следовательно, вполне возможно, хотя и мало вероятно, что, изменив условия, используемые в процессе синтеза аммиака, можно получить гидразин. Однако ни один из этих методов не послужил основой для промышленного способа получения гидразина, главным образом вследствие того, что для практических целей наблюдающиеся выходы слишком низки.
Получение из нитрамида ( NH 2 NO 2).
Нитрамид, являющийся несколько более устойчивым изомером азотноватистой кислоты, может быть восстановлен до гидразина в кислом растворе цинковой пылью. Большой интерес представляют методы, включающие ![]() восстановление ацилпроизводных нитрамида, в особенности таких соединений, как нитромочевина и нитрогуанидин, поскольку не исключено, что эти методы могуг быть использованы в промышленности. Указанные вещества восстанавливаются до соответствующих производных гидразина, а именно до семикарбазида и аминогуанидина, в результате гидролиза которых может быть получен гидразин.
восстановление ацилпроизводных нитрамида, в особенности таких соединений, как нитромочевина и нитрогуанидин, поскольку не исключено, что эти методы могуг быть использованы в промышленности. Указанные вещества восстанавливаются до соответствующих производных гидразина, а именно до семикарбазида и аминогуанидина, в результате гидролиза которых может быть получен гидразин.
![]() NH2NO2N2Н4
NH2NO2N2Н4
NH2CONHNO2![]() +6[Н]
+6[Н] ![]()
![]() NH2СОN2Н3+2Н2O
NH2СОN2Н3+2Н2O
NH2С(NH)NHNO2 NH2С(NH)(N2Н3)
Получение из азотоводородной кислоты и азидов
Гидразин также был получен восстановлением азидной группы. Восстановление может протекать как в щелочном, так и в кислом растворе под действием различных химических агентов, например амальгамы натрия, цинка и соляной или серной кислоты, сульфида натрия или гидроокиси двухвалентного железа. Эти реагенты дают главным образом аммиак и азот и только в очень небольших количествах гидразин. Большой выход получается только в том случае, если гидразин немедленно удаляется из сферы реакции при помощи некоторых методов, например осаждения в форме такой слабо растворимой двойной соли, как сульфат цинка — сульфат гидразина. При восстановлении азотоводородной кислоты в кислом растворе цинком в отсутствие катализатора получаются тем большие выходы гидразина, чем выше температура.
В результате электролиза водных растворов азидов с использованием различных металлических анодов образуется главным образом азот наряду с некоторым количеством аммиака и гидразина. Выходы относительно малы.
Рассмотрим различные стороны реакции Шмидта. Азотоводородная кислота и азиды служат источником имидного радикала, который может реагировать со вторым таким же радикалом, как растворителя, так и растворенных веществ.
NH3 [NH] + H2.
Если раствор азида водорода в бензоле оставить стоять с концентрированной серной кислотой при 15°С, то наряду с такими соединениями, как анилин и гидроксиламин, в сернокислотном слое образуется некоторое количество гидразина.
Выход гидразина заметно понижается с повышением температуры. При 60°С получается только анилин. Было доказано, что имидный радикал, соединяясь с другим таким же радикалом, образует диимид, который затем диспропорционируется, причем получаются азот и гидразин.
2[НN]N2Н2; 2[N2Н2]N2Н4 + N2.
Вероятность такого механизма следует из того факта, что калиевая соль азодикарбоксиловой кислоты подвергается гидролизу в кислом растворе, образуя азот и гидразин, а также азотоводородную кислоту, окись углерода и аммиак. В некотором отношении эту реакцию можно рассматривать как процесс самоокисления производного диимида.
Сульфат гидразина был получен при поглощении безводного азида водорода концентрированной серной кислотой. Механизм этой реакции не был точно выяснен; однако и в этом случае можно сделать предположение о промежуточном образовании радикала NH.
1.2 Разложение аммиака
При полном разложении аммиака образуются водород и азот. Однако было точно установлено, что эти вещества не получаются на первой стадии процесса разложения. Экспериментально была показана возможность побочных реакций с образованием промежуточных продуктов, одним из которых является гидразин. По мнению Рашига, первая стадия разложения аммиака протекает с образованием радикала NН. Можно предположить, что этот радикал способен далее вступать в реакцию конденсации с аммиаком, в соответствии со следующим уравнением:
N3 — NH+2[Н],
NН+NН3 [НNNН3] N2Н4.
Cчитают, что одним из промежуточных продуктов разложения аммиака является радикал NН2 и что он может соединяться со вторым таким же радикалом, образуя гидразин.
NH3 NH2 + [Н]; Н2N+NH2 N2Н4.
Существенным является то обстоятельство, что экспериментальные методы и условия, используемые при разложении аммиака, приводят к еще более быстрому разложению гидразина. Для того чтобы экспериментально продемонстрировать образование гидразина как промежуточного или побочного продукта, необходимо немедленно удалить гидразин из сферы реакции или быстро охладить это соединение, чтобы повысить его устойчивость. Разложение аммиака было осуществлено путем пиролиза, фотохимически в результате фотосенсибилизации, действием электрического разряда на газообразный аммиак и при бомбардировке электронами.
1.3 Синтез Рашига
Очевидно, что успешное использование любого процесса окисления или любого процесса разложения аммиака зависит от быстроты удаления гидразина из сферы реакции. Легкость, с которой сам гидразин подвергается окислению или разложению, затрудняет успешное применение такого рода методов. Единственный процесс промышленного получения гидразина основан на окислении аммиака или его производных гипохлоритом.
Вскоре после открытия гидразина, было установлено что гидразин или гидроксиламин могут быть получены при действии гипохлорита на аммиак. Рашиг доказал, что такое предположение является вполне обоснованным. Исследования Рашига, посвящены одному из наиболее интересных вопросов в области химии азота. Рашиг случайно заметил, что в растворах, содержащих гипохлорит и аммиак, образуется соединение, имеющее свойства восстановителя; это наблюдение привело к разработке метода, который носит имя Рашига. Эмпирически изменяя условия синтеза, Рашиг, наконец, пришел к следующим выводам:
а) избыток аммиака облегчает образование гидразина и
б) образование гидразина из аммиака и хлорамина протекает быстрее при более высоких температурах. Пытаясь определить причину значительных различий в выходах, Рашиг прибегал к добавлению большого числа различных веществ, рассчитывая, что они могут оказывать каталитическое действие.
Им было найдено, что для увеличения выхода гидразина пригодными являются клей и желатина, которые применяются и до настоящего времени. Сначала Рашиг предположил, что эти катализаторы приводят к увеличению вязкости раствора и что образование гидразина легче протекает в более вязкой среде. Последующие исследования, показали, что этот вывод неправилен и что различие в полученных выходах обусловлено действием присутствующих в реакционной смеси ионов металлов, которые катализируют реакцию между гидразином и хлорамином, еще содержащимися в растворе. Клей и желатина способствуют удалению из раствора следов ионов металлов или же препятствуют их вредному действию.
В своих ранних исследованиях Рашиг обнаружил, что гипохлорит натрия и аммиак сначала реагируют с образованием хлорамина. Он показал, что эта реакция протекает довольно быстро и может быть выражена уравнением (1):
NaOCl+ NH3 NH2Cl+ NaOH.(1)
Синтез гидразина, по данным Рашига, обусловлен действием избытка аммиака на хлорамин в соответствии с уравнением (2).
NH2Сl+ NH3 + NaОН N2Н4 + NaСl+ Н2O.(2)
Реакция (2) протекает медленно; она конкурирует с реакцией (3), которая возникает, осложняя процесс. Реакция (3) протекает значительно быстрее; она особенно чувствительна к действию некоторых катализаторов и очень сильно снижает выходы гидразина. Реакция (3) может быть изображена уравнением
2NH2Сl+ N2Н4 2NН4Сl+ N2.(3)
Было найдено, что добавление белковых веществ, например, клея, желатины и альбумина, заметно препятствует реакции (3) и способствует реакции (2), приводя, следовательно, к получению более удовлетворительных выходов гидразина.
Синтез Рашига был с исчерпывающей полнотой изучен многими исследователями с целью определения наилучших условий для достижения максимального выхода. Интерес к этому методу привел к исследованию хлорамина, нахождению оптимальных соотношений между аммиаком и гипохлоритом, обеспечивающих максимальный выход, изучению влияния катализаторов (ингибиторов) и их концентраций на выход гидразина, а также температурных условий, при которых происходит смешивание и протекают последующие реакции.
2.СТРОЕНИЕ МОЛЕКУЛЫ И ДИПОЛЬНЫЙ МОМЕНТ
Большой дипольный момент гидразина (1,83—1,90 дебая) связан с некоторыми очень интересными вопросами, касающимися его строения. В принципе возможно несколько различных структур, отличающихся друг от друга положением атомов водорода по отношению к оси азот — азот в молекуле гидразина. Гидразин можно рассматривать как производное аммиака, в котором вместо одного из атомов водорода находится второй атом азота, расположенный в той же плоскости, что и три атома водорода молекулы аммиака. При этом получается симметричная структура, в которой противоположные моменты связей N—Н должны компенсировать друг друга и давать суммарный дипольный момент, равный нулю. Такая симметричная структура является маловероятной, о чем свидетельствуют как высокий дипольный момент гидразина, так и данные, полученные при изучении этого вопроса, в особенности результаты исследования инфракрасного спектра гидразина. Сначала предполагали, что имеется возможность свободного вращения вокруг оси азот—азот, благодаря чему может существовать любая из возможных форм; считалось также, что большой дипольный момент является результатом равновесия, которое устанавливается между этими предельными структурами. Более поздние исследования N-замещенных гидразина, особенно фенилгидразина и других арилзамещенных, показывают, что эти вещества также характеризуются относительно большими дипольными моментами. Эти дополнительные исследования заставляют предположить, что вращение вокруг оси азот — азот ограничено, если оно вообще возможно. Поэтому симметричная транс-форма маловероятна. Если вращение ограничено, то большой дипольный момент может быть объяснен только конфигурацией, соответствующей изображенной на рис. 2 цис-форме.
Если связи N—Н расположены в пространстве так, как это указано на рисунке, то очевидно, что цис-форма гидразина должна иметь два стереоизомера. Эти выводы подтверждают предположение, впервые высказанное Пенни и Сазерлендом, которые вычислили, что дипольный момент структуры, соответствующей несимметричной цис-форме, равен 1,70 дебая; они предположили также, что валентные углы N—N—Н и Н—N—Н составляют приблизительно 110°С. Электронографические исследования паров гидразина показывают, что углы Н—N—Н и Н—N—N приблизительно составляют 108±10°С. Межатомные расстояния равны:
rN-H=1,04 ± 0,06Е и rN-N= 1,47 ± 0,02 Е. Эти значения валентных углов и межатомных расстояний очень близки к соответствующим значениям для молекулы аммиака. Возможно также, что гидразин существует в таутомерной аминоимидной форме, Н3N NН, и что протон при этом способен мигрировать, образуя молекулу с указанной структурой.
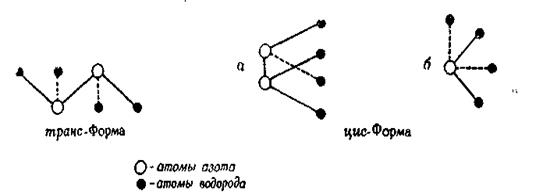 |
Рис. 1. Структуры гидразина.
а—в перспективе; б— ось N-Nперпендикулярна к плоскости рисунка.
Возросший интерес к гидразину и его производным обусловлен отчасти использованием некоторых гидразинов в военной технике [и космических исследованиях] в качестве ракетных топлив, а также разнообразным применением производных гидразина в медицине и сельском хозяйстве.
Гидразин—весьма реакционноспособное соединение: он окисляется на воздухе, окисление протекает через промежуточное образование диимида, давая азот. Как уже отмечалось, превращение гидразина в элементарный азот сопровождается выделением большого количества энергии. Поэтому, а также в результате легкости его получения по методу Рашига гидразин нашел широкое применение в. качестве ракетного топлива. Если использовать его в сочетании с азотной кислотой как окисляющим агентом, то газообразные продукты окисления гидразина (азот, окислы азота) развивают очень эффективную тягу. Некоторыми недостатками гидразина как топлива являются высокая температура плавления, малая стабильность на воздухе и коррозионная активность, затрудняющие хранение и работу с ним.
Из трехфтористого азота при повышенной температуре был получен тетрафторгидразин, но, как и ожидалось, наличие сильно электроотрицательных атомов фтора делает это соединение еще менее стабильным, чем гидразин. Метилгидразин, превосходя гидразин по некоторым физическим показателям, по-видимому, вытеснит последний как жидкое ракетное топливо.
Производные гидразина можно разделить на моно-, ди-, три- и тетразамещенные:
RNH-NH2 RNH-NHR 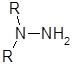

1 2а 2б 3 4
Дизамещенные гидразины 2 следует подразделить на два класса и рассматривать их отдельно, так как первичная аминная функция в 1,1-дизамещенных гидразинах 2б обусловливает свойства, которыми не обладают 1,2-дизамеш.енные гидразины 2а.
Был описан удобный метод аминирования вторичных и третичных аминов до гидразинов и гидразиниевых солей О-гидроксиламинсульфокислотой:
NH 2 O S О3H + R 2 NH —- R 2 N—NH 2 + H 2S О4
Этот реагент является удобным источником частиц NH2 и может найти в будущем более широкое применение.
Сильно нуклеофильный характер гидразина и алкилгидразинов проявляется в различных реакциях. Так, ряд активированных ароматических галогенпроизводных можно ввести в реакцию с гидразином, в результате образуются арилгидразины:
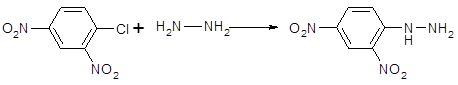
Аналогично гидразин атакует олефины, обедненные электронами, например б,в-ненасыщенные сложные эфиры, с последующей циклизацией в пиразолидоны:
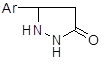 |
![]() ArCH = CHCOOR + H2N—NH2 ArCHCH2COOR
ArCH = CHCOOR + H2N—NH2 ArCHCH2COOR
NH — NH2
Интересный вариант приведенной выше реакции был найден при взаимодействии 1, 1-диалкилгидразинов и акролеина. Здесь начальная атака более нуклеофильного трехзамещен-пого атома азота с последующей циклизацией приводит к четвертичной пиразолиниевой соли. Мягкое разложение этой соли щелочью разрывает связь N—N, давая в-аминонитрил :
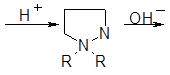 |
R2N – NH2 + CH2=CHCHOR2NCH2CH2CN
Первой стадией этой реакции является образование диметилгидразона, циклизующегося в кислой среде. Последняя стадия представляет собой один из частных случаев аминонитрильной перегруппировки четвертичных альдогидразониевых структур под действием щелочей.
МОНОЗАМЕЩЕННЫЕ ГИДРАЗИНЫ
Монозамещенные гидразины 1 по химическим свойствам подобны незамещенному родоначальнику и также легко окисляются многими окислителями, включая воздух. Другие окислители легко реагируют с монозамещенными гидразинами; так, бром окисляет фенилгидразин до бромбензола и азота. Алкилирование монозамещенных гидразинов дает 1, 1-диалкил- и более замещенные гидразины. Фенилгидразин алкилируется по первому замещенному атому азота (1), хотя многие утверждали, что он метилируется йодистым метилом по второму атому азота, образуя 1-фенил-2-метилгидразин.
Следует отметить, что из возможных переходных состояний при алкилировании замещенного гидразина возникающий положительный заряд будет больше стабилизирован при замещенном атоме азота; промежуточное соединение уравнения (1) стабилизировано индуктивным эффектом ароматического кольца, что невозможно в альтернативном 1,2-ди-замещенном промежуточном соединении:
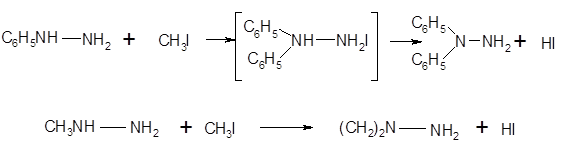 |
(1)
(2)
Монозамещенные гидразины реагируют с различными карбонильными соединениями. В реакции с альдегидами или кетонами продуктами будут гидразоны и вода. С карбоновыми кислотами, хлорангидридами или сложными эфирами образуются 1-замещенные гидразиды .
Из многих гидразинов типа 1 фенилгидразин нашел применение в химии углеводов (например, образование озазонов), а 2,4-динитрофенилгидразин широко используется при идентификации альдегидов и кетонов в виде твердых динитрофенилгидразонов.
ДИЗАМЕЩЕННЫЕ ГИДРАЗИНЫ
1,2-Дизамещенные гидразины
Отщепление азота при окислении 1,2-дизамещенных гидразинов 2а включает две стадии, и промежуточное соединение часто можно выделить, особенно когда R или R (или же оба) — ароматические группы:
RNH—NHR RN = NR R—R + N2
В этой реакции были использованы различные окислители, включая окись ртути, хлорное железо и перманганат калия. Некоторые 1,2-дизамещенные гидразины, особенно те, в которых гидразинный фрагмент заключен в циклическую структуру, окисляются до соответствующих азосоединений при стоянии на воздухе. Многие из промежуточных азосоединений были выделены и затем разложены при нагревании или на свету до азота и углеводородов. Природа заместителей R и R в этих азосоединениях определяет их устойчивость. Если R и R— простые алкильные группы, для выделения азота требуется повышенная температура; некоторые циклические и бензилзамещенные азосоединения разлагаются при комнатной температуре; ароматические же азосоединения вполне устойчивы, 1,2-Диалкилгидразины реагируют с алифатическими альдегидами, давая 1,3, 4-оксадиазолидины, которые можно превратить в 1,2,4-триазолидипы реакцией с первичным амином:
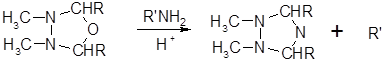
Соединения с двумя атомами азота, связанными через метиленовую группу, можно рассматривать как 1,2-дизамещенные гидразины. Было показано, что трехчленные циклические гидразины можно легко приготовить общим методом из хлорамина и азометинов:

Диазиридины растворяются в органических растворителях, медленно реагируют с кислотой, устойчивы к щелочи и нагреванию (до 100° С). Альтернативным синтетическим методом может служить присоединение реактивов Гриньяра к диазиринам (трехчленным циклическим азосоединениям) с образованием N-алкилдиазиридинов, последние могут дальше гидролизоваться в алкилгидразины:
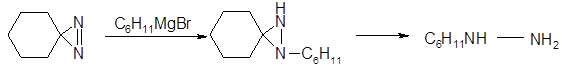 |
B аналогичной реакции 2-метилдиазирина с этилмагнийбромидом получается 1-этил-2-метилдиазиридин, который может также быть приготовлен из хлорамина и этилиденэтиламина:

Известны многие другие примеры циклических гидразинов, в которых оба атома азота включены в цикл. О синтезе четырехчленных циклических гидразинов сведений мало, известна, например, реакция активированных олефинов с диэтил-азокарбоксилатом:

Под влиянием кислот ароматические 1,2-дизамещенные гидразины подвергаются перегруппировкам типа бензидиновой. Механизм этих реакций был предметом интенсивных исследований, и, по-видимому, он включает образование «протонного сандвича»:
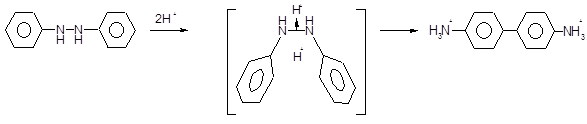
Изучалось поведение 1,2-диалкилгидразинов в условиях реакции Манниха. Реакция гидрохлоридов 1, 2-дизамещенных гидразинов с формальдегидом и ацетофеноном приводит к 3-фенил-1,2-диалкил-Д3-пиразолинам:
 С 6Н 5 С О С Н 3 + (CH 2O)x + RHN—NHR • НС1 [C6H5COCH2 CH2 NR-NHR]
С 6Н 5 С О С Н 3 + (CH 2O)x + RHN—NHR • НС1 [C6H5COCH2 CH2 NR-NHR]
1,1-Дизамещенные гидразины
Благодаря первичной аминной функции 1, 1-дизамещенные гидразины 2б способны к некоторым реакциям, невозможным у изомерных 1,2-дизамещенных гидразинов. Окисление 1,1-дизамещенных гидразинов может привести к двум продуктам и, вероятно, протекает через промежуточный N-нитрен:

Сочетание двух частиц нитрена или, что более вероятно, реакция нитрена с непрореагировавшим гидразином (по типу а) дает тетразены. Наблюдалось также разложение нитрена с образованием азота и углеводорода (по типу б). Продукты окисления зависят от природы заместителей R и R, однако обычно окисление дает тетразен. В некоторых случаях окисление 1,1-дизамещенных гидразинов приводит непосредственно к выделению азота и образованию углеводородо. Этот последний путь, называемый «аномальным окислением», требует, чтобы замещающие группы могли стабилизировать промежуточные фрагменты, образующие новую углерод-углеродную связь. Этими свойствами обладают такие группы, как бензнльная и цианометиленовая:
![]() C6H5CH2NCH2C6H5 + HgO С6Н5СН2СН2С6Н5 + N2 + Hg + Н2О
C6H5CH2NCH2C6H5 + HgO С6Н5СН2СН2С6Н5 + N2 + Hg + Н2О
NH2
Такие аномальные реакции окисления легко происходят в гетерогенных окисляющих системах (например, спирт — окись ртути) с большой поверхностью окислителя, увеличивающей скорость выделения азота. Аналогичные результаты были получены с гетерогенными системами и в других реакциях с выделением азота. Гомогенная среда часто благоприятствует образованию тетразена. Например, в методе получения тетрабензилтетразена из дибензилгидразина используется спиртовой раствор ацетата ртути.
Твердая поверхность, по-видимому, способствует снижению энергии активации выделения азота и, вероятно, помогает изолировать промежуточные частицы N-нитрена друг от друга, препятствуя образованию тетразена. Если в 1, 1-диза-мещенном гидразине присутствуют метальные, этильные, циклогексильные или подобные алкильпые группы, то неспособность этих групп стабилизировать промежуточные углеродные фрагменты (ионы или свободные радикалы — еще не выяснено) подавляет разрыв связей углерод—азот. В этих случаях даже в гетерогенной среде образование азота невыгодно энергетически, и вместо этого получается тетразен.
Такие соединения, как 1-бензил-1-бутилгидразин, окисляются окисью ртути в хлористом метилене, давая с низким выходом амилбензол; однако в этанольном растворе та же реакция приводит к соответствующему тетразену. Другими примерами аномального окисления могут служить:
окисление N-аминоизоиндолина до бензоциклобутена и 1, 2-диметилен-З, 5-циклогексадиена
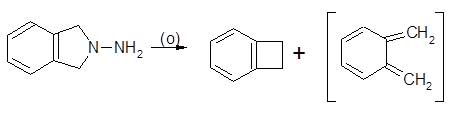
окисление N-амино-1, 3-дифенилизоиндолина в 1,2-дифе-нилбензоциклобутен
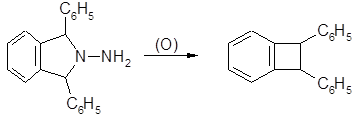
(транс) (транс 81%)
Реакцией, аналогичной аномальному окислению гидразинов 2б, является щелочное разложение сульфонилгидразинов. Эти реакции, по-видимому, протекают через нитрен и продукты часто идентичны продуктам окисления соответствующих гидразинов:

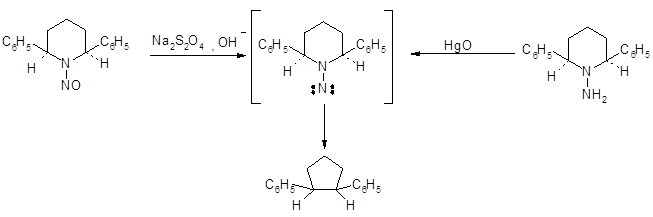
Здесь, как и в реакциях аномального окисления, для образования азота и углеводорода желательны стабилизирующие заместители (например, бензил). Аналогично, обработка некоторых нитрозаминов гидросульфитом натрия в щелочной среде дает азот и углеводороды. Так как продукты реакции идентичны получаемым при окислении соответствующего гидразина окисью ртути, то промежуточное образование нитрена Предполагалось и для этой реакции:
ТРИ- И ТЕТРАЗАМЕЩЕННЫЕ ГИДРАЗИНЫ
Характерной особенностью алифатических трехзамещенных гидразинов 3 является гладкое окисление (при стоянии на воздухе) в дизамещенные гидразоны:
R2N—NHCHR’2 R2N-N = CR’2
Тетразамещенным гидразинам 4 уделяется по сравнению с другими классами меньше внимания. Тетрафенилгндразин, однако, интересен проявлением особых свойств из-за перекрывания объемистых фенильных групп, окружающих маленькие атомы азота. Даже при температуре жидкого воздуха тетрафенилгидразин разлагается в присутствии кислоты с образованием ионов и, возможно, радикалов. Очевидно, в этом соединении связь азот — азот ослаблена вследствие стерического перекрывания, так как другие тетразамещенные гидразины совершенно устойчивы по отношению к кислотам:
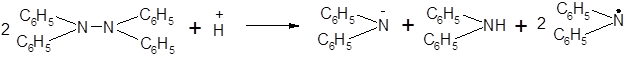 |
Для сочетания двух атомов азота с образованием 1,1-бис-азиридина был использован циклический хлорамин, полученный из этиленимина, что иллюстрирует возможный путь синтеза других тетразамещенных гидразинов:
 |
Получившийся в ходе этой реакции гидразин — слабое основание и разлагается при нагревании в присутствии кислорода со взрывом.
ГИДРАЗИНИЕВЫЕ СОЛИ
Раствор гидразина в воде обладает основными свойствами. Электрометрическое титрование такого раствора показывает, что гидразин является слабым основанием и ведет себя практически как монокислотное основание.
 Были вычислены константы ионизации гидразина при 25°С, которые имеют следующие значения:
Были вычислены константы ионизации гидразина при 25°С, которые имеют следующие значения:
K1 = = 8.5*10-7
 |
K2= = 8.9*10-16
Величина второй константы ионизации настолько мала, что двузамещенные соли гидразина в водном растворе не существуют. Ионы N2H6+ + полностью реагируют с водой (растворителем) в соответствии с уравнением:
![]()
Поскольку гидразин практически является монокислотным основанием, он напоминает скорее аммиак и амины, чем органические диамины. Однако гидразин является значительно более слабым основанием, чем аммиак, что можно видеть при сопоставлении соответствующих констант ионизации, а также теплот нейтрализации этих оснований кислотами в водном растворе.
Гидразин образует не только одно- и двухосновные соли типа N2Н4-2НА, где НА представляет собой обычную одноосновную кислоту, но дает также соединения типа (N2Н4)2Н2В, N2Н4-Н2В иN2Н4-2Н2В. где Н2В — двухосновная кислота. Известны также различные двойные соли, наиболее важными из которых являются двойные сульфаты и двойные хлориды. Кроме того, были исследованы двойные бромиды, иодиды, цианиды ,тиосульфаты , сульфиты , селенаты , нитраты и ферронианиды. Эти двойные соли в основном были получены взаимодействием в водном растворе соответствующих солей металлов и гидразина, взятых в требуемых молярных соотношениях.
Ранее для приготовления простых солей гидразина приходилось применять различные сложные методы. Однако после того как стал доступен водный раствор гидразина, соли легко и быстро можно получать простой нейтрализацией его соответствующими кислотами. В некоторых специальных случаях для этой цели могут быть использованы также реакции двойного разложения с применением сульфата гидразина и соответствующих бариевых солей.
ОБЩИЕ СВОЙСТВА СОЛЕЙ ГИДРАЗИНА
Соли гидразина, содержащие одну молекулу двухосновной кислоты, устойчивы в водных растворах. Однако двухкислотные соли существуют только в твердом состоянии и при растворении в воде немедленно гидролизуются. Так, например, раствор дигидрохлорида гидразина не отличается от раствора, содержащего эквимолекулярные количества свободной хлористоводородной кислоты и моногидрохлорида гидразина.
ы2н4.нх^ад++х-,
Ы2Н4-2НХ+Н20 — ад++2Х-+Н(Н20) + .
Однокислотные соли 1Ч2Н4-НА обычно более устойчивы в водном растворе, чем двухкислотные соединения 1Ч2Н4-2НА. Последние легко могут быть получены кристаллизацией из водных растворов, содержащих избыток кислоты. Поскольку соли гидразина являются аналогами соответствующих солей аммония, то нет ничего неожиданного в том, что большинство из них очень хорошо растворимо в воде и довольно плохо растворимо в неполярных органических растворителях.
Нагревание солей гидразина в большинстве случаев приводит к их разложению; лишь очень немногие из них устойчивы при температурах плавления. Двухкислотные соли при нагревании обычно разлагаются с образованием в качестве промежуточных продуктов однокислотных солей:
К2Н4-2НаД- К2Н4-НА+НА.
Моноалкилгидразины атакуются простейшими галогеналкилами по замещенному атому азота, если не возникает пространственных препятствий. Когда гидразин реагирует с избытком простого галогеналкила (например, йодистого метила), получается 1, 1, 1-тризамещенный гидразин — гидразиниевая соль. Большие алкильные группы, такие как изопропил, не могут размещаться по три у одного атома азота и обычно образуют смеси моно-, 1,1- и 1,2-дизамещенных гидразинов. Лучшим способом получения гидразиниевых солей, допускающим большее разнообразие заместителей, является обработка третичного амина хлорамином:
R3N + NH2C1 R3N+—NH2Cl-
Обычные электронодонорные свойства алкильных групп привели некоторых исследователей к предсказанию более высокой основности (и, следовательно, более низкой кислотности протонированной формы) замещенных гидразинов по сравнению с самим гидразином.
Однако, вопреки ожиданию, метилирование гидразина не повышает основности; гидрохлорид гидразина — более слабая кислота (рКа = 7,95; 8,1), чем гидрохлорид метилгидразина (рКа = 7,87), который в свою очередь слабее гидрохлорида диметилгидразина как кислоты (рКа = 7,21). Такую, кажущуюся аномальной, кислотность гидрохлоридов гидразинов можно объяснить следующим образом: в протонированной форме 1,1-диметилгидразина первый атом азота имеет тетраэдрическую конфигурацию, которая вызывает стерическое отталкивание между метильными группами и вторым атомом азота. Потеря протона и, следовательно, потеря тетраэдрической конфигурации способствует уменьшению напряжения в молекуле. Так как напряжение тетраэдрической конфигурации в диметилгидразине больше, чем в метилгидразине, и еще больше, чем в самом гидразине, потеря протона в случае гидрохлорида диметилгидразина энергетически более выгодна, чем в случае гидрохлоридов метилгидразина или незамещенного гидразина. Равновесие смещается, поэтому вправо, и это увеличивает кислотность гидрохлорида диметилгидразина:
![]()
![]() (CH3)2NH-NH2 (CH3)2N-NH2 + Н+
(CH3)2NH-NH2 (CH3)2N-NH2 + Н+
Так как экспериментальные факты показывают, что дальнейшее алкилирование алкилгидразинов идет по замещенному атому, можно сделать заключение, что замещенный атом азота менее основен, но более нуклеофилен, чем незамещенный.
Аномальную основность алкилгидразинов можно объяснить иначе, связывая ее с гидратацией их в водных растворах по обоим атомам азота и потерей воды при протонировании:
 |
Замещение водорода у любого атома азота на алкильную группу с этой точки зрения должно способствовать гидратации, как реакции, конкурирующей с протонированием, и приводить к снижению рКа (т.е. уменьшению основности аннулируя прямое влияние идуктивного эффекта заместителей на основность. В предпочтенье, что несимметричные полнозамещенные гидразины протокпруютсл по азоту, имеющему большее ihcjoалкидных групп, удается коррелировать рК, с константами Тафта яаместчтглей. Однако кзучгггне основности недавно синтезированных фтор-замешенных моноалкилгидразкков и других гидразинов с электроотрицательными заместителями показало37, что лучшая корреляция получается в предположении ггротсннровання незачещенного атома азота. Проблема основности гидразинов остается, таким образом, достаточно запутанной.
ВОССТАНОВИТЕЛЬНАЯ СПОСОБНОСТЬ ГИДРАЗИНА
Восстановительную способность пробовали использовать в различных системах [см. указанную в списке литературы монографию А. П. Грекова]. Имеются подробные обзоры по восстановлению гидразином олефиновых соединений в присутствии кислорода=-4-а833. В зтой системе, которая особенно удобна для восстановления ненасыщенной части олефиновых кислот, необходимым компонентом является кислород, вводимый энергичным перемешиванием на воздухе. Оптимальные выходы достигаются при 50е С и в узком интервале основности; в атмосфере чистого азота восстановление не идет.
Предполагаемое образование в системе гидразин—кислород промежуточного диимида нашло серьезное подтверждение в работах других исследователей с аналогичными системами4041. При использовании систем гидразин — окислитель было установлено, что непредельные соединения могут восстанавливаться с ««-присоединением водородных атомов, вероятно через циклический промежуточный комплекс (П-30):
| н ч1 нч | н,н 4 С N |
|
| 11 + II — | Г -II | ~ 1 |
| .С А / | W | .С N /Гчн/ |
,сн 1 |
| н | н | н |
Реакция (11-31) иллюстрирует стереоспецифнчность этого метода:
CsH5Cs=CCOOH — CsH5CH = CHCOOH (в основном ч«Ј-форма) (Н-31)
Было показано, что следы ионов, меди оказывают ускоряющее действие на эти процессы. Селективность действия на двойные связи диимида, получаемого разными методами, иллюстрируется его способностью восстанавливать диаллил-дисульфид в дипропилдисульфид.
Доказательство образования диимида из гидразина было получено при масс-спектрографическом изучении электрического разряда в гидразине42. Способность восстанавливать олефиновые связи в мягких условиях, а также в щелочных растворах указывает, что при любых реакциях с гидразином н присутствии кислорода следует принимать в расчет воэчюкное восстановление олефяна в результате образования нестойкого диимида. Это затруднение можно обойти, проводя восстановление в атмосфере азота.
В некоторых 1,2-дизамещенных гидразинах наблюдалось юсетановителъное расщепление связи азот — азот сильными восстановителями (например, гндразобензол расщепляется до анилина).
Основательно было изучено действие никеля Ренея на гидразин43. В таких мягких условиях. как кипячение в мета-юле, эта смесь вызывает разрыв N—N-связи в различных гидразинах и \,Ы-диацилгидразйнах (до амидов).
[Расщепление связи N—N наблюдалось в таких условиях лишь у ароматических гидразосоединений с аминогруппой в пара-положении^45 или в сильно разбавленных спиртовых р-створ ах гидразингидрата 46.
Азобензол и другие ароматические азосоединения гладко восстанавливаются гидразингидратом в присутствии никеля Ренея до гидразосоединений48,47. Недавно было покаадно, что алифатические азосоединения также гладко гидрируются до 1,2-диалкилгидразинов 14.]
Эта же восстановительная система была использована для превращения нитросоединений в амины 484*. Сообщалось 50 об удобном способе получения первичных аминов из нитрилов также при применении гидразингидрата и никеля Ренея в спирте:
RC = N + NH2—NH2 - яаке- Ре:,-Я RCH2NH2 (И-32)
+ N2
П-30)