Огнестойкие композиции на основе полибутилентерефталата
СОДЕРЖАНИЕ: Анализ возможностей повышения огнестойкости вторичного полиэтилентерефталата (ПЭТФ) введением в него в качестве антипирена органоглины. Сущность современных физико-химических методов анализа полимерных материалов. Механизм действия полимерных материалов.Введение
В химии нет отходов, есть только неиспользованное сырье Д. И. Менделеев.
Как известно, развитие современной техники невозможно без исследования пластических масс, в особенности полимерных материалов с пониженной горючестью. Трудносгораемые и трудновоспламеняемые полимеры находят широкое применение в строительстве, машиностроении, электротехнике, авиа- и космической технике, быту.
Пожары, обусловленные воспламенением и горением полимерных материалов, ежегодно наносят большой ущерб различным отраслям экономики. При этом важно отметить, что горение полимеров сопровождается процессами, загрязняющими окружающую природную среду.
В настоящее время эффективным методом снижения горючести полимерных материалов является применение огнегасящих добавок - антипиренов (АП). Но большинство из них в процессе горения полимерных материалов образуют токсичные вещества, наносящие вред человеку и окружающей среде. В связи с этим актуальной является проблема понижения горючести полимеров эффективными и экологически чистыми системами-антипиренами. При этом важным является отказ от широко применяемых, но токсически небезопасных галогенсодержащих соединений, окислов сурьмы и т.д.
Общей тенденцией в данной области исследования являются также вопросы совместимости добавок с полимерами, влияние их на прочностные свойства и технологичность, окраску материалов, а также разработка целевых добавок для конкретных типов полимерных материалов.
Повсеместное использование полимеров также приводит к накоплению отслуживших свой срок пластмассовых изделий в виде длительно не разлагающегося мусора, что приводит к ухудшению экологической обстановки.
Более чем за 10 лет массового потребления в России напитков в упаковке из ПЭТФ на полигонах твердых бытовых отходов накопилось не менее 2 млн. тонн использованной пластиковой тары, являющейся ценным химическим сырьём. Проблема вторичной переработки полиэтилентерефталата (ПЭТФ) является актуальной.
Исходя из этого целью настоящей работы является исследование возможности повышения огнестойкости вторичного ПЭТФ введением в него в качестве антипирена органоглины.
Для достижения поставленной цели были определены следующие задачи:
анализ научной литературы, посвященной данной теме;
изучение современных физико-химических методов анализа полимерных материалов;
проведение экспериментальных исследований;
анализ полученных результатов исследования.
Работа состоит из трех глав, выводов и списка используемой литературы. Первая глава посвящена вопросам снижения горючести полимерных материалов, основным антипиренам, механизму их действия. Во второй главе рассмотрены экспериментальные методики, используемые для изучения свойств полученных композитов. Третья глава работы посвящена обсуждению полученных в ходе исследования результатов.
Глава 1. Литературный обзор
1.1 Горение полимерных материалов
Горение полимерных материалов представляет собой очень сложный физико-химический процесс, включающий как химические реакции деструкции, сшивания и карбонизации полимера в конденсированной фазе, так и физические процессы интенсивных тепло- и массопередачи. Для характеристики негорючих полимеров и материалов с пониженной горючестью пользуются следующими основными терминами:
воспламеняемость - способность материала загореться при определенных условиях (концентрация окислителя, температура и давление окружающей среды). Она характеризуется температурой воспламенения, кислородным индексом и временем зажигания материала;
S горючесть - свойство материала поддерживать горение при окружающих условиях;
S огнестойкость — способность материала сохранять свои свойства в условиях пожара в течение длительного времени;
пожароопасность - степень риска для жизни людей и животных. Под этим термином подразумевают горючесть материала, вероятность его механического разрушения под действием огня и механических нагрузок и выделение токсичных газов и дымов из материала в условиях пожара [1].
Согласно стандартам (например, ГОСТ 28157-89) трудногорючим пластиком считается такой материал, который может загореться при поднесении пламени, но при удалении источника горения самостоятельно затухает через определенный, вполне небольшой, промежуток времени. Существует следующая классификация трудногорючих полимеров. Категория V-0 по UL-94 или ПВ-0 по ГОСТ 28157-89 означает, что при удалении источника пламени стандартный образец, горит не более 10 сек, затем горение самостоятельно прекращается, суммарное время горения 5 образцов должно быть не более 45 сек. Для категории V-1 (ПВ-1) время горения без источника пламени допускается не более 30 сек. Для категории V-2 (ПВ-2) время горения также 30 сек, кроме того, допускается падение горящих капель расплавленного полимера, способных поджечь горючий полимер под образцом [2].
1.2 Влияние строения полимеров на их горючесть
Влияние химического строения полимеров на их горючесть. Процессы горения полимерных материалов обусловлены в основном видом материала, его составом, химическим строением, надмолекулярной структурой. Обычно выделяют начальные химические процессы, связанные с разложением слабых связей, окислением углеводородных группировок, образованием углерод-углеродных связей и элемент-кислородных связей с последующим образованием газообразных и конденсированных продуктов. Любой материал можно характеризовать содержанием горючего, понимая под этим содержание способных к распаду и термоокислению связей. Тогда вероятность погасания материала будет возрастать с увеличением возможности образования при тепловых ударах или воздействии пламени связей, более устойчивых к термоокислению, чем исходные. Обычно энергия связей С-О выше энергий углерод-углеродных связей, поэтому представляет интерес выделение процессов окисления на поверхности материалов. Полиолефины, в которых высока доля связей, способных окисляться, имеют наиболее низкий кислородный индекс (17,5 %), поликарбонаты же можно воспламенить только при содержании кислорода выше чем в атмосфере (26,0 %). Предполагается, что действие регуляторов рения в основном проявляется на начальных стадиях горения [3-5].
Такое деление не всегда оправдывается. Например, некоторые полимеры с разветвленными цепями или содержащие циклические группировки, относят к сгораемым, также к сгораемым относят некоторые сетчатые и трехмерные полимеры. Правда, среди этих полимеров есть такие, которые коксуются на воздухе. Рассмотрим влияние на скорость горения химической природы полимерных материалов. В ряде работ [6-10] указывается па возможность корреляции между химическим строением полимера и способностью его к воспламенению. Отмечено, что уменьшение числа углеводородных группировок приводит к существенному снижению его горючести. Отсюда сделаны выводы о целесообразности снижения воспламеняемости введением в полимеры фрагментов, содержащих конденсированные ароматические кольца.
В работе Ван Кревелена [11, 12] установлена эмпирическая зависимость между кислородными индексами, характеризующими содержание кислорода в азотно-кислородной смеси, достаточное для воспламенения и устойчивого горения полимеров, и содержанием различных инкрементов, составляющих макромолекул полимеров. Аналогичная зависимость найдена для коксовых остатков соответствующих полимеров. Увеличение содержания углеводородных групп соответствует росту количества горючего вещества в полимере, однако при недостаточном потоке окислителя у поверхности, когда скорость термического разложения больше скорости термоокислительного разложения, возможно образование предвестников кокса или сажи [13]. Тогда большое значение начинает приобретать химическое строение углеводородных фрагментов. Например, образование ненасыщенных связей этиленового, ацетиленового или аллильного типа, как известно [14], приводит к появлению ароматических колец или конденсированных ароматических колец. Наличие в полимере ароматических колец способствует в дальнейшем образованию графитоподобных веществ на поверхности. В полимерах одного класса, отличающихся одним или несколькими химическими фрагментами, можно определить влияние строения на горючесть полимеров.
Энергия связи и горючесть полимеров. Между теплотами сгорания, теплотами образования и энергиями связей существует функциональная зависимость [6]. Теплоты сгорания, кислородные индексы и показатели возгораемости взаимосвязаны. Для трудносгораемых полимеров удельные теплоты сгорания составляют менее 21,0 МДж/кг. Для остальных полимеров удельные теплоты сгорания выше указанного, причем отличить по их значениям сгораемые полимеры от трудносгораемых практически невозможно. Например, самозатухающие, судя по кислородным индексам, полигексаметиленадипамид и полихлоропрен выделяют при сгорании столько же или даже больше тепла, чем сгораемые полиметилметакрилат и полиэтилентерефталат. Правда, если сравнить теплоты сгорания, приходящие на связь, то для первых двух полимеров они больше, чем для полиметилметакрилата и полиэтилентерефталата.
Для приближенной оценки затрат на разрушение связи в полимерах в сочетании с удельной теплотой сгорания можно использовать энергоемкость (q) средней связи:
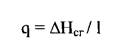
где АНСГ - теплота сгорания полимера, 1 - число связей в полимере.
Энергоемкость средней связи симбатно меняется с изменением энергии средней связи. Обычно для сопоставления термостабильности и в ряде случаев огнестойкости полимеров непосредственно используют значения энергии связей. Однако сопоставление суммы энергии связей с горючестью полимера может принести успех лишь при сочетании у полимеpa термостабильности и термостойкости. С увеличением содержания прочных связей C-F в полифторолефинах повышается значение кислородного индекса и уменьшается показатель возгораемости К.
При наличии в полимерах связей С=О, О-Н, Р=0, S=0, C=N, Si-O, B=N, P=N, энергия которых велика, горючесть полимеров снижается. Введение в полимеры ароматических колец может снизить горючесть полимера и повысить предел огнестойкости. Некоторые трудносгораемые полимеры, например, содержащие галогены или фосфор, не являются термостабильными из-за разрушения связей С-С1, С-Вг, группировок Р-О-С. Пониженная горючесть в этих полимерах обусловлена процессами, ин-гибирующими в поверхностной и предпламенной зонах воспламенение и развитие горения.
Пользуясь значениями средних энергий связей, трудно дать даже приближенную характеристику предлагаемым тепловым свойствам, так как в реальных полимерных телах энергии связей существенно зависят от окружения этих связей. Кроме того, нередко при разрушении полимеров значительную роль играют процессы термического гидролиза, окисления, которые, так же как термическое разложение, оказывают влияние на процессы массо- и теплопереноса.
Влияние некоторых физических параметров полимеров на их горючесть [6, 15]. Горение большинства полимеров, как указывалось выше, является гетерогенным, диффузионным процессом. При этом следует отметить, что диффузионные процессы играют более важную роль, чем химическая активация пиролиза. Это заключение основано на том, что значения кислородных индексов не зависят от химического состава полимера при повышении температуры окружающей среды. Существенное влияние на диффузионные процессы оказывает физическая структура материала или полимера и такие свойства, как плотность, кристалличность, анизотропность, растворимость, набухаемость, газопроницаемость и другие, которые являются проявлениями физической структуры. Физическая структура обусловлена химическим строением полимера, его составом и способом получения, она зависит от сил межмолекулярного взаимодействия и представляет собой наиболее выгодное по плотности упаковки образование макромолекул в данных условиях.
В работе [16] выявлена связь физической структуры полимеров с их горючестью и коксуемостью. Установлено, что при обычных условиях графитизации полиакрилонитрила надмолекулярная структура и ориентация макромолекулярных цепей сохраняется в широком интервале температур (от 20 до 2800 °С) вплоть до образования углеродного материала. Следует отметить, что анизотропия (текстура) в полимерах сохраняется при графитизации в «переходных формах» углерода и в углистом остатке после карбонизации полимера. В качестве примера сохранения текстуры материала можно привести процессы получения углеродных материалов из фенолформальдегидных смол, некоторых полиимидов [6].
Процессы получения углеродных материалов обычно проводят в атмосфере инертного газа при ступенчатом повышении температуры, однако исключить вероятность протекания аналогичных процессов в зоне пиролиза при горении, особенно в том случае, когда материал содержит группировки, способствующие коксованию. Например [17], при наличии в полимерных материалах борфосфор-, фосформеталл- и фосфорсодержащих группировок резко увеличивается выход коксового остатка при линейном пиролизе или горении. Кроме того, эти группировки способствуют формированию упорядоченных форм углерода в условиях карбонизации и графитизации.
Фосфорсодержащие трехмерные полиэфиры с упорядоченной структурой проявляют большую стойкость к огню, чем их аморфные аналоги [6]. При наличии кристалличности, анизотропии в полимерах плотность их повышается, что существенно влияет на горючесть полимерных материалов. Кроме того, увеличение числа сшивок в трехмерных полимерах повышает горючесть.
Следует отметить, что энергия когезии некоторых группировок полимерных макромолекул также играет важную роль в огнестойкости материала.
Так, в ряду галогенсодержащих групп энергия когезии уменьшается при переходе от Вг к С1 и от С1 к F, что соответствует изменению горючести в этом ряду. В частности, присутствие брома в полимере более эффективно содействует уменьшению горючести, чем такое же количество хлора или фтора. Аналогичные сопоставления можно провести между энергиями когезии и коксовыми числами полимеров. Из этих сравнений следует, что при уменьшении содержания метиленовых групп или при введении вместо них ароматических, амид-ных, аминных, гидроксильных, сложноэфирных или галогенсодержащих групп коксовые числа увеличиваются [6].
Учитывая сказанное о влиянии физической структуры на процессы коксования, симбатное изменение энергии когезии и коксовых чисел можно легко объяснить.
Наличие в полимерах таких гетероатомов, как фосфор, бор, барий, кальций, способствует, как уже указывалось, структурированию и увеличению выхода кокса. Объясняют это образованием на поверхности материалов минеральных поверхностных слоев. Например, высокий коксовый остаток полиарилатов, содержащих в цепи карборановые группы, обусловлен образованием минеральной пленки брутто-формулы В203. Считают, что аналогичные защитные пленки образуются на поверхности материала, содержащего фосфор или металл [18, 19]. Это является причиной снижения их горючести.
1.3 Способы снижения горючести полимерных материалов
Методы снижения горючести полимерных материалов основаны на следующих принципах:
изменение теплового баланса пламени за счет увеличения различного рода теплопотерь;
снижение потока тепла от пламени на полимер за счет создания защитных слоев, например образующего кокса;
уменьшение скорости газификации полимера;
изменение соотношения горючих и негорючих продуктов разложения материала в пользу негорючих [9].
Существует несколько способов снижения горючести полимерных материалов, которые можно условно разделить на следующие группы:
огнезащита с использованием устойчивых к пламени материалов (огнезащитных покрытий);
введение наполнителей горения или антипирирующих составов;
модификация полимерных материалов.
Наряду с первым и вторым способами используют пропитку полимерных материалов огнегасящими составами, способными образовывать на поверхности материала защитный слой.
Огнезащита устойчивыми к пламени материалами подразумевает покрытие плитками, листами из негорючих или трудносгораемых материалов изделий из горючих материалов. В качестве огнезащитных покрытий могут применяться огнезащитные краски, лаки, вспенивающие покрытия. Преимущества огнезащитных покрытий - в простоте изготовления и сравнительно небольшой стоимости работ. Недостатком этого способа является то, что при повышении температуры огнезащитное покрытие отслаивается от основного горючего материала, что вызывает загорание основного материала. Для вспенивающихся покрытий, на которых при воздействии огня или тепла образуется быстрорастущая негорючая пена с мелкими закрытыми порами, снижение адгезии покрытия к материалу менее вероятно из-за резкого уменьшения теплопередачи через покрытие.
Введение наполнителей приводит к некоторому снижению горючести, некоторые замедлите горения (красный фосфор, Sb203, соли фосфорной кислоты и т.д.) можно рассматривать как наполнители, в том случае, когда не наблюдается их растворение в материале. В качестве армирующих материалов широко применяют стекловолокна, асбест, углеродные волокна, улучшающие физико-механические характеристики, теплостойкость и вместе с тем приводящие к снижению горючести полимеров [20].
В качестве порошкообразных наполнителей, способствующих снижению горючести, применяют окислы и гидроокиси некоторых металлов, графит, окислы кремния (SiC^), сурьмы (Sb203), бораты цинка (Zn3(B03)2), природные неорганические вещества (каолин, пемза, гипс, перлит, монтмориллонит, вермикулит), различные соли, такие как оксалаты и карбонаты [21]. Многие из перечисленных порошков являются ингибиторами воспламенения и горения и находят применение в качестве огнетушащих веществ. Из ингибиторов горения в пламенной зоне наиболее эффективны окислы, затем в порядке уменьшения эффективности следуют соли: карбонаты, бромиды, сульфаты и фосфаты.
Широкое применение для строительных негорючих полимерных материалов различного назначения получили такие наполнители, как песок, перлит, вермикулит и окись кремния. Каолин, мел, гидроокись алюминия, мелкодисперсный карбонат кальция применяют при изготовлении резин.
На горючесть наполненных полимерных материалов оказывает влияние не только химическая природа наполнителя, но и дисперсность, а также прочность сцепления наполнителя и связующего. С увеличением адгезии возрастает прочность, что зачастую сопровождается увеличением огнестойкости и стабильности к термоокислению. Однако даже в случае удачного подбора наполнителя процесс воспламенения и горения композиционных полимерных материалов определяется степенью однородности и изотропности материала, концентрацией негорючих частиц в поверхностном слое материала.
Немалую роль в снижении горючести материалов при введении наполнителей играет степень наполнения. Например, в результате увеличения содержания связующего в минераловатных плитах с 4 до 8 % изменяется группа возгораемости материала: сгораемые плиты становятся трудносгораемыми. Преимущества введения наполнителей - одновременное улучшение ряда эксплуатационных характеристик материала. Основной недостаток при этом заключается в том, что при повышенных температурах происходит расслаивание материала.
Введение замедлителей горения и составов, замедляющих горение, в полимерные материалы заключается обычно в равномерном распределении этих веществ - антипирена в объеме материала. Этот способ более эффективен по сравнению с предыдущим из-за термических превращений замедлителей горе ния в зоне пиролиза и поверхностной зоне, а также диффузии продуктов их превращений на поверхности материала. При этом концентрация продуктов термических превращений замедлителей горения в поверхностной зоне резко возрастает, что в свою очередь ведет к ускорению коксования материала. Основным недостатком этого способа является в ряде случаев увеличение горючести материалов в процессе его эксплуатации, поскольку введенные замедлители горения могут «выпотевать», вымываться или иным способом выделяться из материала. В свою очередь все эти факты будут способствовать загрязнению окружающей природной среды [20, 22].
Физические методы снижения горючести полимеров. Проблему снижения горючести полимерных материалов следует рассматривать, принимая во внимание многостадийный характер процесса их диффузионного горения. К числу физических методов понижения горючести можно отнести следующие [6]:
замедление подвода тепла к полимерному материалу;
охлаждение зон горения в результате увеличения физических стоков тепла в окружающую среду. Так, эффективным является отток тепла от полимерного покрытия через теплопроводящую подложку, потеря на испарение компонентов, унос тепла расплавленными каплями;
ухудшение условий переноса реагентов к области горения. В данном случае создают физические барьеры между полимером и окисляющейся средой, замедляют диффузию горючих компонентов в композитах;
срыв пламени потоком газа;
воздействие акустического, гравитационного полей и т. д.
Один из эффективных физических методов повышения огнестойкости полимерных материалов огнезащита [23]; для огнезащиты используют устойчивые к пламени материалы - плитки, листы из негорючих или трудносгораемых материалов. В качестве огнезащитных покрытий могут применяться огнезащитные краски, лаки, вспенивающие покрытия.
Покрытия, наносимые на поверхность защищаемого материала можно разделить на 3 группы:
трудновоспламеняемые и негорючие покрытия;
теплоизолирующие негорючие покрытия;
наиболее перспективные покрытия вспучивающего типа. При горении они вспучиваются, образуя карбонизированный пенообразный слой, низкая теплопроводность которого защищает объект от теплового потока пламени.
Огнезащитные покрытия вспучивающего типа включают несколько компонентов разного назначения: а) вещества, являющиеся источником углеродного каркаса: б) вещества, катализирующие реакции образования углеродного скелета: в) вспенивающие агенты [24].
Химические методы регулирования горения. К химическому модифицированию полимеров можно отнести использование реакционноспособных антипиренов, которые включаются в молекулярную структуру конечного продукта в результате совместных полиреакций с исходным мономером [21].
Применение антипиренов - наиболее распространенный эффективный способ снижения горючести полимерных материалов. Наряду с реакционноспособными антипиренами широко используют антипирены аддитивного типа. Они механически совмещаются с полимерным субстратом [25].
Подбор замедлителей горения проводят эмпирическим путем по факторам, указывающим на снижение горючести материала. К этим факторам относятся:
Образование негорючих газов, которые уменьшают содержание горючих компонентов в газовой смеси, а также вероятность контакта кислорода воздуха с нагретой поверхностью материала.
Эндотермическое разложение самих веществ, замедляющих горение.
Деструкция замедлителей горения с образованием акцепторов свободных радикалов, которые взаимодействуют с продуктами цепных реакции в пламени.
Образование прочного кокса или оксидной пленки или негорючего пенного слоя на поверхности материала, которые уменьшают перенос тепла от пламени к материалу и предотвращают воздействие активных частиц пламени и кислорода воздуха на полимерные материалы.
Образование высоко дисперсных частиц, которые уменьшают распространение пламени и изменение направления химических реакций, приводит к образованию менее реакционноспособных радикалов.
Указанные факторы являются результатом процессов, протекающих в зоне пиролиза и поверхностном слое материала. Применение замедлителей горения эффективно, если они способствуют:
S образованию графитоподобных веществ;
S получению на поверхности материала негорючей углеродной пены с закрытыми порами;
возникновению в поверхностных слоях материала парамагнитных центров, прекращающих цепные реакции распада материала, или частиц, активных молекул, ингибирующих горение материала в предпламенной зоне.
При подборе замедлителей горения или антипирирующих составов для различных полимерных материалов необходимо проводить комплексное исследование свойств самих замедлителей горения и антипирирующих составов с учетом изменения свойств этих материалов в процессе термических превращений данных веществ. Кроме того, необходимо знать поведение полученных огнестойких материалов в процессе эксплуатации при действии экстремальных тепловых нагрузок или при горении [26, 27].
1.4 Замедление дымообразования
Процесс дымообразования при горении полимерных материалов очень важен с точки загрязнения окружающей природной среды. Так, образование сажистого дыма является наиболее типичным при горении полимерных материалов. Кроме этого при горении полимерных материалов в атмосферу поступают различные продукты горения, которые зачастую бывают вредные и токсичные. В свою очередь образование сажистого дыма означает неполноту сгорания органической составляющей. Антипирены, позволяющие снизить горючесть полимерных материалов, чаще всего приводят к увеличению их сажеобразующей способности. Достаточно трудно одновременно снизить горючесть и сажеобразование при горении полимерных материалов. Подавление сажеобразования при горении газовых систем осуществляется путем изменения соотношения между топливом и окислителем, аэродинамических условий потоков, а именно увеличения скорости окислителя. При этом реализуется более полное сгорание горючего. Аналогична ситуация и в случае большинства полимерных материалов.
В случае полимерных систем образование дыма зависит, прежде всего, от условий выделения летучих продуктов и их состава. Влияние различных факторов на дымообразование полимеров показано в работе [28].
Для снижения дымообразования используют различные соединения металлов: цианид и тиоцианат меди, окись железа, смеси порошкообразного железа с окисью меди или молибдена, смеси окисей меди, молибдена и ванадия. Эффективно также использовать кислородсодержащие добавки кислотного типа.
Обогащение кислородсодержащими веществами летучих продуктов уменьшает их сажеобразующую способность в результате не только появления источника дополнительного кислорода в пламени, но и склонности кислородсодержащих соединений к захвату электрона и образованию отрицательных ионов. Рекомбинация последних с положительными углеводородными ионами - зародышами сажевых частиц - снижает вероятность энуклеации сажи.
При применении металлсодержащих веществ возможны альтернативные воздействия: а) добавки влияют на пиролиз полимера таким образом, что изменяется состав летучих или растет выход коксовых остатков; б) добавки в результате превращений в конденсированной фазе образуют при горении соединений летучие, которые переходят в газовую фазу и подавляют сажеобразование.
1.5 Основные антипирены, применяемые для повышения огнестойкости полимеров
Антипирены (АП) - это вещества, которые влияют на химию процессов в конденсированной или газовой фазе, или на поверхности раздела фаз. Антипирены препятствуют горению полимерных материалов и относятся к важнейшим компонентам пластмасс. При горении полимерных материалов внутри и на поверхности конденсированной фазы происходят сложные физико-химические процессы, в результате которых полимер превращается в нагретые до высокой температуры продукты сгорания.
Предохраняющее действие антипиренов определяется:
низкой температурой их плавления с образованием плотной плёнки, преграждающей доступ кислорода к материалу;
разложением антипиренов при нагревании с выделением инертных газов или паров, затрудняющих воспламенение газообразных продуктов разложения предохраняемого материала;
поглощением большого количества теплоты на плавление, испарение и диссоциацию антипиренов, что предохраняет пропитанные материалы от нагревания до температуры их разложения;
повышенным углеобразованием пропитанных материалов при их термическом разложении за счёт образующихся кислот.
В подавляющем большинстве воздействие антипиренов на горение полимерных материалов является множественным. В структуре антипирена могут одновременно присутствовать элементы пламегасящего действия и группы, которые способны оказывать влияние на ход пиролиза полимеров и гетерогенное окисление.
Эффективные замедлители горения, действие которых проявляется в зоне пиролиза в поверхностном слое, должны способствовать образованию коксового слоя на 80 % поверхности материала. При слоистом строении замедлителей горения или огнезамедлительных систем и хемосорбции на этих слоях макромолекул полимера, как представлено ранее, возможно образование «заготовок» углеродного слоя, процентное содержание которых равно процентному содержанию закоксованной поверхности.
Рассмотрим механизм ингибирования реакций в пламени в присутствии различных добавок: а) галогенсодержащие органические соединения
Для снижения горючести полимерных материалов чаще всего применяют галогенсодержащие соединения [8, 9, 29]. Они бывают трех типов: соединения с алифатической или циклоалифатической структурой. Природа и число атомов галогена в каждом типе структуры варьируется.
В качестве антипиренов используют низко- и высокомолекулярные соединения. Широко применяется декабромдифенилоксид (ДБД), гексабромциклододекан (ГБЦД), которые снижают температуру горения за счет протекания сложного комплекса радикальных реакций (механизм см. ниже). Они практически полностью заменили в рецептурах трудногорючих композитов устаревшие виды антипиренов - гексабромбензол, хлорпарафин. ГБЦД более эффективен, особенно в полистирольных пластиках, но применение его ограничено из-за низкой температуры разложения. Однако его термостойкость можно поднять до 220 - 23 0 °С, стабилизируя ГБДЦ стеаратами тяжелых металлов. Здесь важно отметить, что данные антипирены с точки зрения экологии уступают достаточно сильно гидроксидам, т.е. они летучи при повышенных температурах.
Эффективность замедления пламенных реакций галогенсодержащими соединениями одинакового строения, различающимися природой галогена, как известно [30], растет в последовательности F CI Вг I.
В зависимости от строения галогенсодержащие соединения подвергаются пиролизу либо в конденсированной фазе, либо испаряются и деструктируют уже в газовой фазе. В свою очередь это обстоятельство приводит к загрязнению окружающей природной среды. В частности, первичные реакции пиролиза галогенсодержащих соединений приводят, как правило, к образованию НХ и RXn, реже - Х2, где X - атом галогена. Однако отщепление НХ от макромолекул алифатической структуры сопровождается чаще всего образованием ненасыщенных систем. Превращения последних в конденсированной фазе обуславливают образование нелетучего карбонизованного остатка, что, в конечном счете, сказывается на скорости горения материала [31].
Зависимость эффективности ингибирующего действия вещества от природы галогена, влияние малых концентраций добавок подтверждают, что механизм ингибирования воспламенения и горения модельных w*— их производными имеет химическую основу. Установлено, что галогены и их соединения не влияют на окисление углерода до окиси углерода.. В то же время они существенно ингибируют окисление СО до С02.
Для объяснения наблюдаемых эффектов ингибирования пламенных режут производными галогенов в свое время были предложены различные механизмы. В их основе заложено участие различных галогенсодержащих молекул, атомов или ионов галогена в разных стадиях радикального цепного процесса горения. Как приводится в работе [3], ингибирование пламени обусловлено реакциями с участием атомов кислорода и образованием промежуточных соединений - оксигалогенов:
![]()
Оксигалогены быстро реагируют с активными центрами Н и ОН, снижая их концентрацию и тормозя, таким образом, скорость процесса окисления.
Некоторые исследователи отводят большую роль в ингибировании пла мени галогенсодержащими соединениями заряженным частицам Х~ [3]. Так, пламени при атаке электрона галогенсодержащие соединения диссоциирую образуя отрицательный ион галогена и радикал:
![]()
соединения металлов
В качестве антипиренов используют соли, окислы, гидроокиси и органические производные металлов [33].
Наиболее широко используемый недорогой антипирен А1(ОН)3 [34, 35]. Его потребление в мире составляет 43 % по объему из всего количества антипиренов. Сдерживает его использование низкая термостойкость - до 190 °С, при более высоких температурах он разлагается с выделением большого количества воды. Для достижения необходимой огнестойкости приходится вводить гидро-ксид алюминия в полимер в больших количествах (50 - 70 %). А1(ОН)3 за счет выделения воды поглощает тепло сгорания, подавляет выделение дыма и снижает долю кислорода в прилегающем к изделию слое. Служит также экономичным наполнителем. Используется главным образом в электроизоляционных и кабельных изделиях на основе ПЭ и сополимеров этилена. Чтобы снизить отрицательное влияние высокого наполнения на физико-химические свойства материала, используют тонкодисперсные поверхностно модифицированные марки А1(ОН)3, обработанные стеариновой кислотой или органосилановыми аппретами.
Гидроксид магния Mg(OH)2 имеет более высокую термостойкость -332°С, используется в тех полимерах, где нужна высокая температура переработки [34, 35]. Более эффективен по сравнению с гидроксидом алюминия, менее абразивен, уровни ввода его ниже. Хотя по западной литературе он считается более выгодным, но в России он дороже, собственное производство отсутствует и используется гидроксид магния значительно реже. Гидроксиды лучше подавляют дымовыделение по сравнению с бромсодержащими антипиренами.
Большинство из них обладают низкой упругостью паров, что исключает присутствие в газовой фазе в нормальных условиях горения. Поэтому механизм их действия чаще всего связан с процессами, протекающими в конденсированной фазе.
Исследование ингибирования углеводородного пламени различными соединениями показало, что некоторые соединения металлов более эффективно замедляют развитие процесса горения, чем галогенсодержащие органические соединения [36]. Эффективность замедления горения зависит не только от природы металла, но и от природы субстрата.
Ингибирующее действие металлсодержащих соединений связывают с участием последних в катализе гибели активных центров (атомов и радикалов), ответственных за развитие цепного процесса горения веществ. При этом осуществляется ли в присутствии соединений металлов гомогенный или гетерогенный механизм гибели активных центров, зависит от многих факторов.
Сторонники гомогенного механизма ингибирующего действия соединений металлов полагают, что последние в пламени испаряются, разлагаются и образуют активные промежуточные формы соединений. Такой активной формой являются, например, гидроокиси металлов. В частности, гидроокиси щелочных металлов легко образуются в пламени при разложении солей металлов в присутствии паров воды. В богатых топливом пламенях взаимодействие щелочных металлов с парами воды (продуктом сгорания) может протекать по равновесной реакции:
![]()
Следует отметить, что в бедных топливом пламенях образование активных промежуточных металлсодержащих частиц осуществляется в результате непосредственного окисления щелочных металлов. При этом образуется надо-кись металла:
![]()
В свою очередь надокись металла легко реагирует с активными центрами процесса горения, образуя более стабильные, но активные соединения:
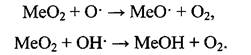
Последние далее реагируют с активными центрами процесса горения.
Главной особенностью монтмориллонита является его способность к адсорбции различных ионов (в основном катионов), а также к ионному обмену. С водой образует пластичные массы, при этом, разбухая, может увеличиваться в объеме в 10 раз. Входит в состав бентонитовых глин (слово «бентонит» - происходит от названия местности Бентон в США) [48].
Неорганические слои глин образуют скопления с зазорами между ними, называемыми прослойками или галереями. Изоморфное замещение внутри слоев (Mg2+ замещает А13+ в октаэдрической или А13+ замещает Si4+ в тетраэдрической структурах) генерирует отрицательные заряды, которые электростатически уравновешиваются катионами щелочных или щёлочноземельных металлов, расположенных в прослойках (рис. 1.1.). Этим обусловлена высокая гидрофильность бентонита. При помещении бентонита в воду, она проникает в межслоевое пространство монтмориллонита, гидратирует его поверхность и обменные катионы, что вызывает набухание минерала. При дальнейшем разбавлении водой бентонит образует устойчивую вязкую суспензию с выраженными тиксотропными свойствами.
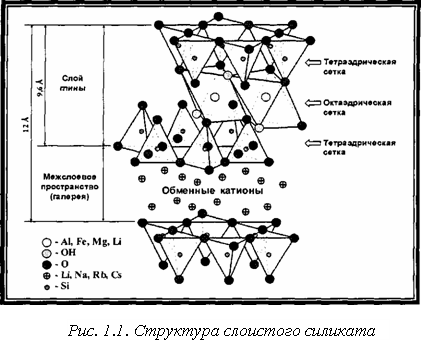
Монтмориллонит обладает высокими катионообменными и адсорбционными свойствами, которые наиболее выражены у бентонитов, монтмориллонит которых содержит преимущественно обменные катионы натрия.
1.6 Способы модификации слоистых силикатов
Слоистые силикаты обладают весьма специфическими свойствами - резким падением прочности при увлажнении, разжижением при динамических воздействиях, набуханием при обводнении и усадки при высушивании.
Гидрофильность алюмосиликатов является причиной их несовместимости с органической полимерной матрицей — это основная проблема, которую приходится преодолевать при создании полимерных композитов. Ее решают путем модификации глины органическим веществом
Модифицированная глина (органоглина) имеет следующие преимущества:
органоглина хорошо диспергируется в полимерной матрице;
органоглина взаимодействует с цепочкой полимера.
Модификация алюмосиликатов может быть осуществлена путем замещения неорганических катионов внутри прослоек органическими катионами. Замещение катионными поверхностно-активными веществами, такими, как объёмные аммоний- и фосфоний-ионы (в данной работе используются гуанидиний-ионы), увеличивает пространство между слоями, уменьшает поверхностную энергию глины и придает поверхности глины гидрофобный характер. Модифицированные вышеуказанным путем глины лучше совмещаются с полимерами и образуют слоисто-полимерные нанокомпозиты [49]. Наряду с ионными органическими модификаторами глин могут быть использованы неионные модификаторы, которые связываются с поверхностью глины за счет водородных связей. В некоторых случаях органоглины, полученные с использованием неионных модификаторов оказываются более химически стабильными, чем органоглины, полученные с использованием катионных модификаторов.
Как правило, наименьшая степень десорбции наблюдается в случае неионного взаимодействия между поверхностью глины и органического модификатора. По всей видимости, водородные связи, образованные между этиленок сидной группой и поверхностью глины делают эти органоглины химически более стабильными, чем органоглины, полученные по ионному механизму.
1.7 Структура полимерных композитов на основе монтмориллонита
Изучение распределения органоглины в полимерной матрице имеет большое значение, так как свойства получаемых композитов напрямую зависят от степени распределения органоглины.
Процесс формирования нанокомпозита протекает через ряд промежуточных стадий (рис. 1.2.) [50]. На первой стадии происходит образование тактоида - полимер окружает агломераты органоглины. На второй стадии происходит проникновение полимера в межслойное пространство органоглины, в результате чего происходит раздвижение слоев до 2-3 нм. Дальнейшее увеличение расстояния между слоями (третья стадия) приводит к частичному расслоению и дезориентации слоев органоглины. Эксфолиация или расслоение наблюдается, когда полимер раздвигает слои глины на 8 - 10 нм и более.
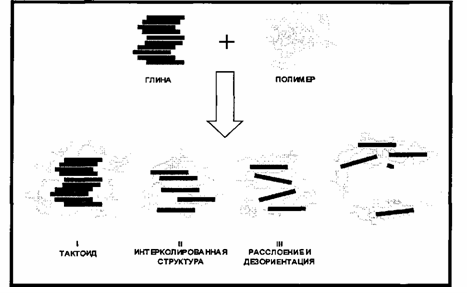
Рис. 1.2. Схема образования полимерного нанокомпозита
На самом деле, в получаемых полимерных композитах могут присутствовать все указанные структуры, что зависит от степени распределения органог-лины в полимерной матрице. Расшелушенная (эксфолиированная) структура является результатом очень хорошей степени распределения органоглины. При избытке органоглины и плохой степени диспергирования возможно присутствие агломератов органоглины в полимерной матрице, что подтверждается методом рентгено-структурного анализа [47].
При изучении полимерных композитов используется ряд специфических методов, которые позволяют судить о структуре материала (рентгено-структурный анализ, сканирующая (СЭМ), трансмиссионная (ТЭМ) электронная микроскопия и др.). Сравнивая данные рентгено-структурного анализа для органоглины и композитов можно определить оптимальное количество глины, которое необходимо вводить в композит.
В зависимости от степени распределения частиц глины в полимере выделяют интеркалированную и эксфолиированную структуру нанокомпозитов (рис. 1.З.). Надо заметить, что хотя на рисунке пластинки глины показаны жесткими, на самом деле они обладают некоторой гибкостью. Формирование интеркалированной или эксфолиированной структуры зависит от многих факторов, например, от способа получения нанокомпозита, от природы глины и т.д. [48].
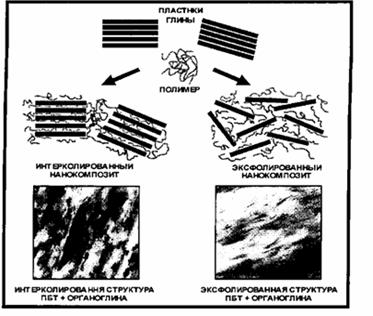
Рис. 1.3. Формирование интеркалированной и эксфолированной
структуры композитов. Если поверхность композита гладкая, то частицы органоглины распределены равномерно. Поверхность композита, обычно, становится деформированной при увеличении содержания органоглины. Возможно, это влияние агломератов глины.
При содержании органоглины 2-3 масс. % слои глины разделены слоем по-ера толщиной ~ 4-10 нм. При большем содержании органоглины 4-5 масс.%
1.8 Способы получения полимерных композитов на основе алюмосиликатов
Разработаны следующие методы получения композитов на основе органоглин:
в процессе синтеза полимера;
в расплаве;
в растворе;
золь-гель процесс [48].
Для получения полимерных композитов на основе органоглин наиболее широко применяются методы получения в расплаве и в процессе синтеза полимера. Получение полимерного композита в процессе синтеза самого полимера заключается в интеркалировании мономера в слои глины. Мономер мигрирует сквозь галереи органоглины, и полимеризация происходит внутри слоев (рис. 1.4.) [51].
Реакция полимеризации может быть инициирована нагреванием, излучением или соответствующим инициатором. Очевидно, что при использовании этого метода должны получаться наиболее удовлетворительные результаты по степени распределения частиц глины в полимерной матрице. Это может быть связано с тем, что раздвижение слоев глины происходит уже в процессе внедрения мономера в межслойное пространство. Это означает, что силой, способствующей расслоению глины, является рост полимерной цепи. В то время как при получении полимерных нанокомпозитов в растворе или расплаве основным фактором достижения необходимой степени распределения глины является лишь удовлетворительное перемешивание. Желательно проводить процесс синтеза нанокомпозита в вакууме или токе инертного газа. Помимо этого, для удовлетворительного диспергирования органоглины в полимерной матрице необходимы большие скорости перемешивания.
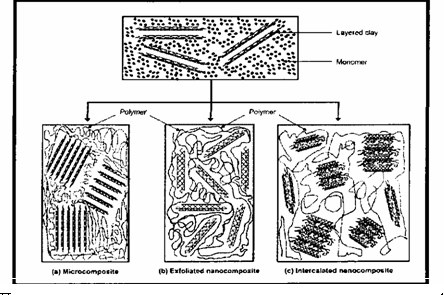
Рис. 1.4. Получение полимерного нанокомпозита в процессе синтеза самого полимера (in situ) (а) - микрокомпозит, (Ь) - эксфолированный (расше-лушенныи) нанокомпозит, (с) - интеркалированный нанокомпозит.
Метод получения полимерных нанокомпозитов в расплаве (экструзион- ный) состоит в смешении расплавленного полимера с органоглиной. В ходе интеркаляции полимерные цепи в существенной степени теряют конформационную энтропию. Вероятной движущей силой для этого процесса является важный вклад энтальпии взаимодействия полимер-органоглина при смешении. Стоит добавить, что полимерные нанокомпозиты на основе органоглин успешно получают экструзией [50]. Преимуществом экструзионного метода является отсутствие каких-либо растворителей, что исключает наличие вредных стоков, скорость процесса значительно выше, более простое технологическое оформление производства. То есть для получения полимерных композитов в промышленных масштабах экструзионный метод является наиболее предпочтительным, требующим меньших затрат на сырьё и обслуживание технологической схемы.
При получении полимер-силикатных нанокомпозитов в растворе органо-силикат набухает в полярном растворителе, таком как толуол или N,N-диметилформамид. Далее к нему добавляется раствор полимера, который про никает в межслоевое пространство силиката. После этого проводится удаление растворителя путем испарения в вакууме. Основное преимущество этого метода заключается в том, что полимер-слоистый силикат может получаться на основе полимера с низкой полярностью или неполярного материала. Тем не менее, этот метод не находит широкого использования в промышленности по причине большого расхода растворителя [49].
При получении нанокомпозитов на основе различной керамики и полимеров применяется золь-гель технология, в которой исходными компонентами служат алкоголяты некоторых элементов и органические олигомеры. Сначала алкоголяты подвергают гидролизу, а затем проводят реакцию поликонденсации гидроксидов. В результате образуется керамика из неорганической трехмерной сетки. Существует также метод синтеза, в котором полимеризация и образование неорганического стекла протекают одновременно. Возможно применение нанокомпозитов на основе керамики и полимеров в качестве специальных твердых защитных покрытий, а также как оптические волокна [52].
1.9 Свойства полимерных композитов
Посредством введения наночастиц органоглины в полимерную матрицу, удается улучшить термическую стабильность и механические свойства полимеров. Достигается это благодаря объединению комплекса свойств органического (легкость, гибкость, пластичность) и неорганического (прочность, теплостойкость, химическая устойчивость) материалов.
Композиты демонстрируют существенное изменение свойств по сравнению с ненаполненными полимерами. Так при введении в полимерную матрицу модифицированных слоистых силикатов в пределах 2-10 вес. % наблюдается изменение: механических свойств, таких как прочность на растяжение, сжатие, изгиб и излом; барьерных свойств, таких, как проницаемость и стойкость к воздействию растворителей; оптических свойств и т.д. Плюс к этому повышается огнестойкость (температуростойкость), ударопрочность и практически отсутствует увеличение веса полимера и физико-механические свойства не ухудшаются как при обычных наполнениях, а существенно улучшаются.
Другие интересные свойства, демонстрируемые композитами полимер-органоглина включают повышенную термостабильность и стойкость к распространению пламени даже при очень низких концентрациях наполнителя. Формирование термоизоляции и незначительная проницаемость обугленного полимера для огня обеспечивают преимущества использования этих материалов.
Использование органоглины в качестве добавки в полимеры может изменять такие свойства, как температура деструкции, огнестойкость, упругость, прочность на разрыв. Важными свойствами композитов являются [53] увеличение модуля упругости, понижение коэффициента термического расширения, низкая газопроницаемость, повышенная устойчивость к действию растворителей. В композитах наблюдается широкий комплекс барьерных свойств.
Глава 2. Экспериментальная часть
2.1 Материалы и методики смешения
В работе в качестве исходных материалов использованы вторичный полиэтилентерефталат (ВПЭТФ), т. е. использованные пластиковые бутылки из-под минеральной воды, и органомодифицированная глина (месторождение Герпегеж). Предварительно собранные бутылки, прежде всего, отмывались от этикеток и других посторонних веществ. Для отмывки использовали моющие средства. Затем вторичный полиэтилентерефталат вручную измельчили в мелкую крошку. После этих операций вторичный ПЭТФ сушили при температуре 100 ± 5 °С в глубоком вакууме в течение двух часов.
Рабочие композиции на основе высушенного вторичного полиэтилентерефталата и органомодифицированной глины готовили следующим образом. Вначале приготовили концентрат на основе ВПЭТФ и органоглины, затем этот концентрат диспергировали в основной массе полимера экструзией на одношнековом экструдере фирмы «Betol» (Великобритания) с диаметром шнека 25 мм. Процесс экструдирования проводили при скорости вращения шнека 70 + 100 об/мин; температуре материального цилиндра 260 °С и температуре формирующей головки 230 °С. Затем экструдаты гранулировали и использовали для изготовления соответствующих образцов для физико-химических исследований. Содержание органоглины во вторичном ПЭТФ варьировали в интервале 0,5 + 5 масс. %.
2.2 Приготовление образцов
Пластины на основе исходного полимера и композитов на его основе и органоглины для оценки огнестойкости с размерами 100x1 Ох 1мм получали методом прессования при температуре 250 °С, а также давлении 250 кгс/см (ГОСТ 25.601 - 80). Фиксация формы изделия происходит в результате охлаждения в пресс-форме до комнатной температуры. Затем на данных пластинах проводились исследования на горючесть.
Образцы для ударных испытаний получали литьем под давлением ~ 10 МПа на термопластавтомате «KuASY-l,6 х 2/1» (Германия) при температуре 260 °С.
2.3 Методика приготовления органоглины
В водную суспензию Са-монтмориллонита, полученную путем перемешивания на магнитной мешалке в течение двух часов, добавляли метакрилат туанидина (МАГ) в концентрации 5 % от массы монтмориллонита (50 г) и перемешивали еще 4 часа. Полученную органоглину промывали дистиллированной водой многократной декантацией. Высушивали при комнатной температуре.
2.4 Методика определения скорости горения
Горение принято характеризовать значениями линейных и массовых скоростей выгорания полимерных материалов. При лабораторных исследованиях определяют время самостоятельного горения материала. Исходя из этого, в на стоящей работе оценку эффективности действия замедлителей горения оценивали по продолжительности самостоятельного горения композитов согласно ГОСТ 21207. 81. Для этого образец, пластинку размером 100x10x1 мм, закрепили по ширине в горизонтальном положении таким образом, чтобы длина незакрепленной части образца была не менее 80 мм. Затем образец поджигали горелкой, пламя которой устанавливается под углом 45 ± 1°. Через 60 сек после поджога образца, горелку выключают, одновременно включают секундомер и измеряют время горения образца.
Метод предназначен для сравнительной оценки относительной способности пластмасс воспламеняться под воздействием источника зажигания.
Время горения определяли под вытяжкой. Образцы поджигали газовой горелкой Бунзена (рис. 2.1.), диаметром (9,5 ± 0,5) мм, с использованием смеси газов: пропан-бутан.
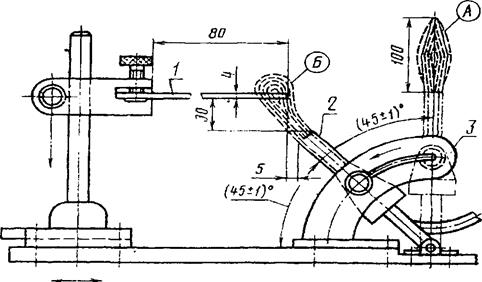
Рис. 2.1. Устройство газовой горелки Бунзена: 1 - образец; 2 - горелка Бунзена; 3 - поворотное приспособление.
2.5 Определение коксового остатка
Коксовый остаток определялся термогравиметрическим методом. Образцы исходного вторичного полиэтилентерефталата и композитов ВПЭТФ + органоглина выдерживали при температуре 800 °С в муфельной печи в течение часа. Скорость подъема температуры 5 °С/мин. Затем находили разницу в массе навесок до и после выдерживания в муфельной печи и вычисляли коксовый остаток (%).
2.6 Измерение показателя текучести расплава (ПТР)
Показатель текучести расплавов (ПТР) или индекс расплава - условная величина, характеризующая поведение полимера в вязкотекучем состоянии при переработке его в изделия методами литья под давлением, экструзии и др. ПТР обычно определяют для термопластичных материалов (полиэтилен, полипропилен, полиформальдегид, ПЭТФ и др.) и выражают количеством полимера в граммах, которое проходит через стандартное сопло в течение 10 мин при определенных температуре и нагрузке.
ПТР, характеризующий реологические свойства расплавов вторичного ЭТФ и его композиций с органоглиной, определяют на капиллярном вискози- метре ИИРТ-М (рис. 2.2.), который представляет собой стальной цилиндрический корпус 4, имеющий два продольных канала. Один канал служит для загрузки.
На верхней части поршня находится втулка 2, на которой помещен съемный груз 1. В нижней части центрального канала укреплено стандартное сопло 6, выполненное из закаленной стали. Сопло не должно выступать за пределы корпуса.
Корпус пластометра имеет электрообогрев 5, при помощи которого можно создавать в цилиндре необходимую температуру. Температура поддерживается автоматически и регулируется при помощи электронного потенциометра.
Прибор снабжен выдавливающим устройством для удаления остатков испытуемого полимера. Все поверхности пластометра, соприкасающиеся с материалом, должны быть отполированы. Пластометр устанавливают вертикально и укрепляют на металлической подставке 7 [67].
Для испытуемого материала с ПТР от 0,15 до 25 г/10 мин применяют
стандартное сопло с внутренним диаметром 2,095 ± 0,005 мм. При большой текучести полимеров (от 25 до 250 г/10 мин) применяется сопло с внутренним диаметром от 1,160 до 1,200 мм.
Полимер для определения ПТР используют в виде порошка или неболь
ших гранул.
Вес груза Р (в г) для стандартного сопла рассчитывают по формуле:
![]()
где, D - диаметр поршня, мм; d - диаметр сопла, мм.
Колебания в весе груза допускаются в пределах ±10 г.
Перед началом испытаний прибор нагревают до необходимой температуры (в данном случае - 260 °С), выдерживают в течение 15 мин, а затем в центральный канал вводят навеску испытуемого полимера и опускают поршень без груза. Спустя 3-4 мин, когда установится необходимая температура полимера,
на поршень помещают груз (в данном случае груз стандартный - 2,160 кг). Материал начинает выдавливаться через сопло пластометра. Первую выдавленную порцию загрузки (примерно 1/3) отбрасывают, а последующие порции срезают через определенные промежутки времени (5 с) и после охлаждения взвешивают.
Расчет ПТР осуществляют по следующей формуле:
М-600 ПТР = где М - масса (усредненная по пяти значениям) полимерного образца, выдавленного из сопла пластометра через каждые 5 с; 600 - стандартное время испытаний для большинства полимеров, с; Т - время опыта, с [67].
В условном обозначении ПТР верхний индекс обозначает температуру испытаний в °С, а нижний - нагрузку в кг, при которой выполнены измерения ПТР.
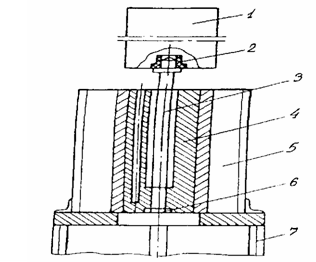
Рис. 2.2. Схема прибора для определения показателя текучести расплава полимера: 1 - груз; 2 - втулка; 3 - поршень; 4 - цилиндр; 5 - электрообогрев; 6 - сопло; 7 — подставка.
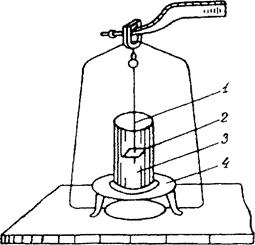
2.7 Измерение плотности
Плотность литых образцов определяют методом гидростатического взвешивания согласно методике [67]. Для этого отлитую таблетку, взвешивают с точностью до 0,002 г. Погружают в жидкость, в которой исходный вторичный ПЭТФ, а также композиции на его основе не растворяются и не набухают), для удаления с поверхности таблетки пузырьков воздуха их вытирают фильтровальной бумагой. После этого образец подвешивают на очень тонкой проволоке к крючку над чашкой весов и подставляют стакан с жидкостью (с дистиллированной водой), в которой проводят определение. Стакан ставят на специальну подставку, которая не должна касаться чашки весов. Образец с проволокой погружают в воду при 20 °С и взвешивают. Затем взвешивают проволоку без образца при этом же уровне погружения. Схема прибора для определения плотно- гидростатическим взвешиванием представлена на рис. 2.3. Плотность полимерных композиций р (г/см3) вычисляют по формуле:
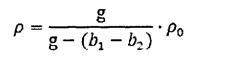
Ударные испытания по методу Шарпи
Ударные испытания выполнены согласно общепринятой методике Шарпи трехточечный высокоскоростной изгиб) - ГОСТ 4746-80, образцы типоразмера, имеющие следующие размеры: длина L = 50 мм, ширина В = 6 мм и толщина = 4 мм,. Ударные испытания выполнены на маятниковом копре ИТ-1/4 со малой энергии 1,0 Дж. Скорость х ударника в момент контакта с образцом равнялась 2,9 м/с (согласно паспорту). Общий вид такой установки показан на рисунке 2.4. [69].
Ударную вязкость Ар для исходного вторичного ПЭТФ и композиций на основе вторичного ПЭТФ и органоглины определяли по формуле:
![]()
где, U - энергия разрушения образца, Дж; В - ширина образца, мм; D - толщина образца композита, мм.
Образцы для ударных испытаний получены литьем под давлением
ЮМПа на термопластавтомате «KuASY-l,6 х 2/1» (Германия) при температуре 260 °С.

Рис. 2.4. Общий вид установки для ударных испытаний по методике Шарпи.
2.9 Статистическая обработка данных
Любые измерения сопровождаются той или иной ошибкой или погрешностью, которые можно разделить на два вида: систематические и случайные. [70]
В ходе исследования физико-химических свойств полимера проводили несколько определений, которые характеризуются воспроизводимостью полученных результатов, зависящей от случайных погрешностей, и правильностью результатов, являющейся следствием систематической погрешности.
Для оценки воспроизводимости результатов эксперимента используем методы математической статистики, разработанные для малого числа измерений п.
Доверительный интервал. При отсутствии систематической погрешности среднее арифметическое значение х не совсем совпадает с истинным значением величины. Отличие носит вероятностный характер и может быть оценено с учетом несовпадения реального t-распределения погрешностей с распределением при бесконечно большом числе определений.
Численное значение ширины доверительного интервала 5 зависит как от числа выполненных определений п, так и от выбранного значения доверительной вероятности Р:
![]()
Где tpin - коэффициент Стьюдента, численные значения которого приводятся в справочной литературе.
Полученные результаты представляют в виде интервального значения определенной величины:
![]()
что равнозначно указанию четырех величин: х, S„, п, Р.
Воспроизводимость измерения выражают также в виде относительной погрешности прямого измерения Ах (%):
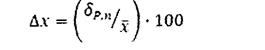
Все измерения следует выполнять с одинаковой относительной недостоверностью [71].
Глава 3. Обсуждение результатов исследования
Как известно, развитие современной техники невозможно без исследования пластических масс в особенности полимерных материалов с пониженной горючестью. Пожары, обусловленные воспламенением и горением полимерных материалов ежегодно наносят большой вред человеку. Во многих странах мира приняты специальные постановления об ограничении использования горючих полимерных материалов в строительстве промышленных и гражданских сооружений, при проектировании и создании транспортных средств, в электротехнике, электронике, производстве товаров бытового назначения. Принятие этих мер способствует интенсификации научных исследований по огнестойким полимерным материалам.
При этом пожарная опасность материалов и изделий из них определяется в технике следующими характеристиками: 1) горючестью, т.е. способностью материала загораться, поддерживать и распространять процесс горения; 2) дымовыделением при горении и воздействии пламени; 3) токсичностью продуктов горения и пиролиза - разложения веществ под действием высоких температур; 4) огнестойкостью конструкции, т.е. степенью сохранения физико-механических и функциональных свойств изделия при воздействии пламени [65]. Практически все полимеры можно условно разделить на 2 большие группы по отношению к тепловому воздействию:
разлагающиеся почти нацело (коксовые числа их не превышают 1-2 %);
карбонизирующиеся при нагревании. Согласно Ван Кревелену [11] достаточно коксовое число полимера довести до 10 %, чтобы кислородный индекс полимера повысился до 21.5 %. При достижении коксового числа 20-25 % полимер попадает в разряд трудногорючего или негорючего.
Эффективным методом понижения горючести полимерных материалов является применение огнегасящих добавок - антипиренов. Антипирены - это вещества, которые влияют на химию процессов в конденсированной или газовой фазе, или на поверхности раздела фаз.
Действие антипиренов проявляется в следующих характеристиках:
изменение состава летучих продуктов пиролиза полимеров;
вменение выхода кокса;
способность выделятся из полимерного субстрата в процессе горения;
зависимость эффекта замедления горения от природы окисли-
теля и структуры полимера.
В случае, если не удается изменить направление реакций термического разложения полимера в сторону образования кокса, наиболее эффективным путем снижения горючести является применение антипиренов газофазного действия.
Универсальных антипиренов для разных полимеров не существует, что объясняется, прежде всего, специфическим взаимодействием полимера с антипиреном, индивидуальными термическими характеристиками полимера и добавок.
Снижение горючести полимерных материалов можно проводить двумя путями: физические и химические меры воздействия на процесс го
рения. Но они используются для подавления уже возникшего процесса горения.
Методы снижения горючести полимерных материалах основаны на таких принципах как:
1)изменение теплового баланса пламени за счет увеличения раз- личного рода теплопотерь;
снижение потока тепла от пламени на полимер за счет создания защитных слоев, например из образующегося кокса;
уменьшение скорости газификации полимера;
изменение соотношения горючих и негорючих продуктов разложения материалов в пользу негорючих.
Учитывая эти принципы, а также экологическую сторону проблемы, достаточно перспективным направлением в области снижения горючести полимерных материалов является, применение в качестве антипиренов органомодифицированные глины. В связи с этим в работе в качестве ан-типирена для вторичного полиэтилентерефталата, использован органомо-дифицированный монтмориллонит. Выбор органоглины еще обусловлен отсутствием их негативного воздействия на окружающую среду, дешевизной и доступностью.
3.1 Оценка горючести композитов ВПЭТФ + органомодифицированный ММТ по скорости горения
Для оценки огнестойкости полимерных материалов нами были использованы такие характеристики как линейная скорость выгорания образцов и коксовый остаток.
Для определения линейной скорости выгорания применяли стандартные пластины с размерами 100x10x1 мм, высота пламени 100 мм.
Как показали наши исследования, горение исходного вторичного ПЭТФ сопровождается достаточно сильным капанием расплава полимера, образованием черного дыма, интенсивным горением. В отличие от него композиты, содержащие органомодифицированный монтмориллонит горят значительно медленнее, с незначительным образованием капель расплава полимера и дыма. Последнее обстоятельство играет важную роль в предотвращении попадания вредных продуктов разложения полимерных материалов в окружающую природную среду. Уменьшение линейной скорости выгорания и незначительное образование капель расплава в композитах ВПЭТФ + органоглина, очевидно, связано со снижением доступа кислорода из воздуха в зону пламени или с образованием коксовой корки органоглины на поверхности полимерного материала. Это образование снижает, во-первых, выход горючих продуктов в газовую фазу, во-вторых, уменьшает поток горючих газов к пламени. В свою очередь эти обстоятельства препятствуют распространению пламени, снижая тем самым скорость горения материала. Действительно, углерод, остающийся в твердой фазе, мог бы попасть в пламя и окислиться до углекислого таза с большим тепловым эффектом. Конечно, в большом пожаре этим все дело и кончится, и никакой пользы от образования кокса мы не получим, разве, что снижение выбросов загрязняющих веществ. Но следует заметить, что в данном случае опасность исходит от слабых источников зажигания, поэтому эффект от образования кокса так важен.
При этом следует отметить, что образующаяся в поверхностной зоне корка ММТ способствует охлаждению поверхностных слоев материала, т. е. температура поверхности композиционного материала при горении ниже температуры пламени. Последнее обстоятельство приводит к большим тепловым потерям, и скорость горения материала становится малой. Кроме того, следует отметить, что корка монтмориллонита будет препятствовать переходу активных различных частиц из материала в пламенную зону тем самым снижая, горючесть данного материала. В результате этого композиты остывают раньше, чем превращаются в газообразные продукты горения.
На рисунке 3.1. приведена зависимость линейной скорости выгорания полученных композитов от содержания антипирена. Как видно из рисунка, введение органоглины способствует уже при концентрации АП 0,5
масс. % значительному снижению горючести полимера. А при введении органомодифицированного монтмориллонита во ВПЭТФ в количестве 2 и 5 масс. % данные композиционные материалы гаснут сразу и через 40 с, соответственно. При этом следует отметить, что горение данных композитов сопровождалось незначительным каплеобразованием и присутствием черного дыма. Однако композиты ВПЭТФ + органоглина по значениям времени линейной скорости выгорания лучше исходного вторичного ПЭТФ.
В полученных композитах вследствие снижения теплового эффекта процесса горения и катализа процесса коксования, количество коксового остатка всегда увеличивается, как будет показано ниже.
Анализ литературных источников показал, что антипирены на основе органоглин считаются более экологически чистыми, чем широко применяемые в промышленности галогенсодержащие соединения, при разложении которых в процессе горения образуются токсические вещества.
Оценка горючести композитов ВПЭТФ + органомодифицированный ММТ по коксовому остатку
Для подтверждения понижения горючести полученных композитов нами был определен коксовый остаток по данным термических исследований.
Анализ результатов термических исследований полученных нами композитов показывает, что введение во вторичный ПЭТФ органоглины способствует увеличению коксового остатка по сравнению с исходным полимером. Причем для композитов на основе ВПЭТФ и органоглины в количестве 1 и 2 масс. % характерно значительное коксовое число 34 %, 38 %, соответственно (рис. 3.2.).
Результаты определения КО согласуются со значениями линейной скорости выгорания композитов. Как видно, из рис. 3.2, добавка органоглины в той или иной мере повышает огнестойкость ВПЭТФ.
Кроме того, важно отметить, что при повышенных температурах органоглина не разлагается с выделением токсичных веществ в окружающую среду. В этом плане органоглина весьма перспективная добавка к полимерным материалам не только для повышения их огнестойкости, но и в качестве экологически чистого антипирена, продукты разложения которой не представляют опасности для людей. Исследования в этом направлении в настоящее время интенсивно ведутся.
Оценка физико-механических свойств композитов ВПЭТФ + органомодифицированный ММТ
Перед исследователями стоит проблема найти оптимальное количество антипирена, так чтобы остальные физико-механические показатели материала не ухудшались бы.
Для анализа влияния органомодифицированного монтмориллонита (антипирена) на эксплуатационные характеристики вторичного ПЭТФ и нахождения оптимальной концентрации, которая не оказывает негативное воздействие на исходный комплекс физико-механических характеристик, нами были проведены реологические и механические исследования полученных композитов.
Информативными и надежными методами оценки реологических и механических свойств полученных композиций являются определение показателя текучести расплава (ПТР) и деформационно-прочностных характеристик.
Характеристикой практической значимости для полимерных материалов служит показатель текучести расплава. По изменению ПТР можно судить о структурных и реологических изменениях, об изменении молекулярной массы полимера, происходящих в полимерной матрице после введения антипиренов.
Можно предположить, что при смешении полимера и органоглины образуется эксфолированная структура. В целом значения ПТР полученных композитов выше, чем у исходного полимера и близки к значениям ПТР промышленного ПЭТФ.
Важным критерием при выборе антипиренов является сохранение механических свойств исходного полимера. С этой целью нами было исследовано влияние органоглины на ударную вязкость ВПЭТФ.
Кроме того, одной из характеристик полимера, на что следует обратить внимание - это надмолекулярная структура. При этом, одним из параметров характеризующих надмолекулярную структуру и во многом определяющей механические свойства, является плотность. Плотность оценивали гидростатическим взвешиванием.