Протеомика
СОДЕРЖАНИЕ: В отличие от многих других наук о протеомике можно точно сказать, при каких обстоятельствах она возникла, указать год, когда появилось ее название и кто его придумал.А.А. ЗАМЯТНИН, доктор биологических наук, Институт биохимии им. А.Н.Баха РАН
Наш рассказ будет посвящен одной из самых молодых фундаментальных наук (если не самой молодой), которая родилась всего лишь несколько лет назад вместе с теми, кто еще сейчас учится в начальной школе. В отличие от многих других наук о протеомике можно точно сказать, при каких обстоятельствах она возникла, указать год, когда появилось ее название и кто его придумал.
Начнем с обстоятельств. Во второй половине XX в. бурно развивались аналитические методы биохимии, молекулярной биологии и вычислительной техники. Выдающиеся успехи, достигнутые в этих областях, привели к возможности расшифровки огромных последовательностей оснований нуклеиновых кислот и к записи полного генома живого организма. Впервые полный геном был расшифрован в 1980 г. [1] у бактериофага phi Х-174 (около 5·103 оснований), затем у первой бактерии – Haemophilus influenzae (1, 8·106 оснований) [2]. А c завершением XX в. была закончена грандиозная работа по расшифровке полного генома человека – выявлению последовательности примерно 3 млрд оснований нуклеиновых кислот [3]. На эту работу было затрачено несколько миллиардов долларов (примерно по одному доллару на одно основание). Всего же уже расшифрованы геномы нескольких десятков видов живых организмов. Именно в этот период возникли две новые биологические науки: в 1987 г. впервые в научной печати было использовано слово «геномика» [4], а в 1993 г. – «биоинформатика» [5].
У каждого биологического вида часть генома представлена участками, кодирующими аминокислотные последовательности белков. Например, таких участков у человека насчитывается порядка 100 000 (по некоторым оценкам, это число может достигать 300 000, а с учетом химически модифицированных структур – нескольких миллионов). Казалось бы, зная полный геном и генетический код, можно путем трансляции получить все сведения о структуре белков. Однако все не так просто. Постепенно становилось очевидным, что в данной рассматриваемой клеточной системе организма нет корреляции между наборами мРНК и белков. Кроме того, многие белки, синтезированные на рибосомах в соответствии с нуклеотидной последовательностью, после синтеза подвергаются химическим модификациям и могут существовать в организме в модифицированной и немодифицированной формах. И еще немаловажно то, что белки обладают разнообразными пространственными структурами, которые на сегодняшний день нельзя определить по линейным последовательностям нуклеотидов и даже аминокислот. Поэтому прямое выделение и определение структур всех функционирующих белков остается по-прежнему актуальной задачей (прямое определение структуры на сегодняшний день осуществлено примерно лишь для 10% белков человека). Так, в дополнение к геномике появился термин «протеомика», объектом исследования которой является протеом (от англ. PROTEins – белки и genOMe – геном). А в научной печати упоминание о протеоме впервые появилось в 1995 г. [6].
Следует добавить, что большую роль в жизнедеятельности организмов играют многочисленные короткие фрагменты белковых предшественников, которые называются олигопептидами, или просто пептидами. Именно из-за них наблюдается такой разнобой в оценке количества белково-пептидных компонентов у представителей одного биологического вида. Поэтому наряду с терминами «протеом» и «протеомика» в настоящее время уже употребляются такие термины, как «пептидом» и «пептидомика», представляющие собой часть протеома и протеомики. О многообразии структуры и функций белков и пептидов на страницах газеты «Биология» нами было рассказано ранее [7].
Итак, сформулируем определения новых наук, которые появились при жизни нынешнего молодого поколения и которые тесно взаимосвязаны друг с другом (рис. 1).
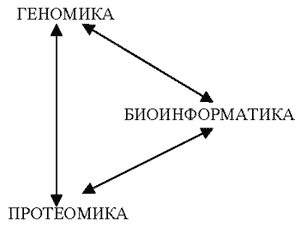
Рис. 1. Схема, иллюстрирующая полную взаимосвязь трех новых биологических наук
Геномика – наука, занимающаяся изучением структуры и функций генов (геном – совокупность всех генов организма).
Биоинформатика – наука, занимающаяся изучением биологической информации с помощью математических, статистических и компьютерных методов.
Протеомика – наука, занимающаяся изучением совокупности белков и их взаимодействий в живых организмах (протеом – совокупность всех белков организма).
Отметим также, что протеомика в общих чертах включает в себя структурную протеомику, функциональную протеомику и прикладную протеомику, которые мы рассмотрим в отдельности.
Структурная протеомика
Наиболее яркой особенностью биологии является разнообразие. Оно просматривается на всех уровнях биологической организации (биологические виды, морфология, химическая структура молекул, сеть регуляторных процессов и т.д.). В полной мере это относится и к белкам. Масштаб их структурного разнообразия до сих пор до конца не выявлен. Достаточно сказать, что число аминокислотных остатков в одном белке может составлять от двух (минимальная структура, имеющая пептидную связь) до десятков тысяч, а белок титин человека содержит 34 350 аминокислотных остатков и на сегодняшний день является рекордсменом – самой крупной из всех известных белковых молекул.
Чтобы получить сведения о протеоме, необходимо сначала его выделить и очистить от других молекул. Поскольку число белков во всем протеоме (т.е. во всем организме) весьма велико, обычно берут только часть организма (его орган или ткань) и различными методами выделяют белковую компоненту. За почти 200-летнюю историю изучения белков разработано множество методов выделения белков – от простого солевого осаждения до современных сложных методов, учитывающих различные физические и химические свойства этих веществ. После получения чистой фракции индивидуального белка определяется его химическая структура.
В структурной протеомике проводится определение структуры не одного, а сразу множества белков, и к настоящему времени для этого разработан специальный цикл процедур и создан арсенал соответствующих высокоточных приборов. (Полный набор оборудования для протеомных исследований стоит более одного миллиона долларов.)
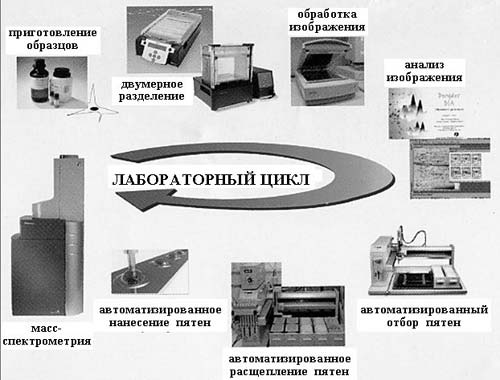
Рис. 2. Инструменты протеомики
На рис. 2 приведена схема лабораторного цикла от приготовления образца до определения его структуры. После выделения и очистки (на рисунке представлен уже выделенный и очищенный препарат) с помощью двумерного электрофореза проводится разделение белков. Это разделение идет по двум направлениям: в одном разделяются молекулы белка, имеющие разную массу, в другом – различный суммарный электрический заряд. В результате этой тончайшей процедуры на специальном носителе одинаковые молекулы группируются, образуя макроскопические пятна, причем в каждом пятне содержатся только одинаковые молекулы. Число пятен, т.е. число разных белков или пептидов, может составлять многие тысячи (рис. 3, 4), и для их исследования используются автоматические устройства для обработки и анализа. Затем проводится отбор пятен и введение содержащихся в них веществ в сложнейший физический прибор – масс-спектрометр, с помощью которого и определяется химическая (первичная) структура каждого белка.
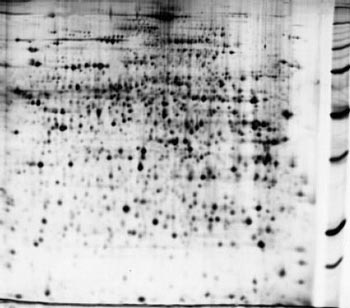
Рис. 3. Пример двумерной электрофореграммы белков из экстракта печени мыши [8]

Рис. 4. Пример двумерной электрофореграммы пептидов из цереброспинальной жидкости человека [9]

Рис. 5. Нуклеотидная последовательность гена, кодирующего сывороточный альбумин человека
Первичную структуру белка можно также определить, пользуясь результатами геномики и биоинформатики. На рис. 5 дана полная структура гена сывороточного альбумина человека. Она содержит 1830 азотистых оснований, кодирующих 610 аминокислотных остатков. Этот ген, как и абсолютное большинство других, начинается с кодона atg, кодирующего остаток метионина, и заканчивается одним из стоп-кодонов, в данном случае taa. Таким образом кодируется структура, состоящая из 609 аминокислотных остатков (рис. 6). Однако эта структура – молекула еще не сывороточного альбумина, а лишь его предшественника. Первые 24 аминокислотных остатка представляют собой так называемый сигнальный пептид, который при переходе молекулы из ядра в цитоплазму отщепляется, и только после этого образуется структура сывороточного альбумина, получаемая при выделении этого белка. В итоге данная молекула содержит 385 аминокислотных остатков.
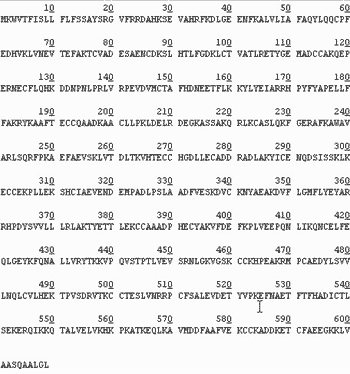
Рис. 6. Аминокислотная последовательность предшественника сывороточного альбумина человека, транслированная с нуклеотидной последовательности с помощью генетического кода
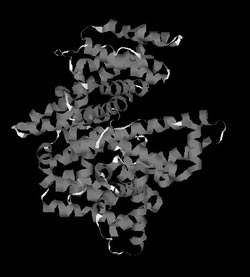
Рис. 7. Пространственная (третичная) структура молекулы сывороточного альбумина человека
Однако аминокислотная последовательность не раскрывает пространственную структуру белка. С точки зрения термодинамики, вытянутая линейная структура энергетически невыгодна, и поэтому она специфическим для каждой последовательности образом сворачивается в уникальную пространственную структуру, которая может быть определена с помощью двух мощных физических методов – рентгеноструктурного анализа и метода ядерного магнитного резонанса (ЯМР-спектроскопии). С помощью первого из них определены пространственные структуры уже нескольких тысяч белков, в том числе и сывороточного альбумина человека, изображение которого представлено на рис. 7. Эта структура, в отличие от первичной (аминокислотной последовательности), называется третичной и в ней хорошо видны спирализованные участки, являющиеся элементами вторичной структуры.
Таким образом, задача структурной протеомики сводится к выделению, очистке, определению первичной, вторичной и третичной структур всех белков живого организма, а ее основными средствами являются двумерный электрофорез, масс-спектрометрия и биоинформатика.
Биоинформатика белков
Существование огромного количества разнообразных белков привело к необходимости создания информационных массивов – баз (или банков) данных, в которые заносились бы все известные о них сведения. В настоящее время существует множество общих и специализированных баз данных, которые доступны в Интернете каждому желающему. В общих базах содержатся сведения о всех известных белках живых организмов, т.е. о глобальном протеоме всего живого. Примером такой базы является SwissProt-TrEMBL (Швейцария–Германия), в которой на сегодняшний день содержатся структуры почти 200 000 белков, установленные аналитическими методами, и еще почти 2 млн структур, которые определены в результате трансляции с нуклеотидных последовательностей [10]. На рис. 8 и 9 показано количество существующих белков, которые известны для каждого заданного числа аминокислотных остатков. Оси абсцисс на этих графиках ограничены 2000 остатков, но, как уже сказано выше, хотя и не часто, но встречаются и существенно более крупные молекулы. Из данных, представленных на рисунках, следует, что наибольшее число белков содержит по несколько сотен аминокислотных остатков. К ним относятся ферменты и другие достаточно мобильные молекулы. Среди более крупных белков много таких, которые выполняют опорную или защитную функции, скрепляя биологические структуры и придавая им прочность.
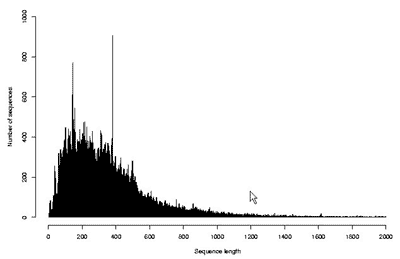
Рис. 8. Распределение известных (выделенных) белков по числу аминокислотных остатков
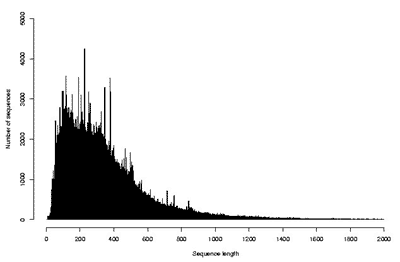
Рис. 9. Распределение транслированных аминокислотных последовательностей по числу минокислотных остатков
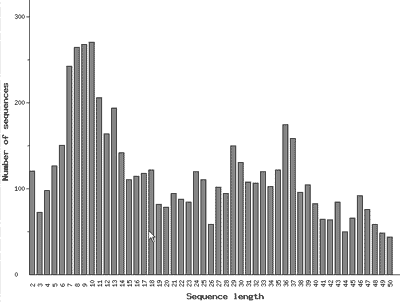
Рис. 10. Распределение известных природных олигопептидов по числу аминокислотных остатков
В глобальном протеоме особое место занимают небольшие очень подвижные молекулы, содержащие не более 50 аминокислотных остатков и обладающие специфическим спектром функциональной активности. Они называются олигопептидами, или просто пептидами. Для них, т.е. для глобального пептидома, создан особый банк данных, который называется EROP-Moscow. Это название представляет собой аббревиатуру от термина Endogenous Regulatory OligoPeptides (эндогенные регуляторные олигопептиды), и указывает на то, что банк создан и базируется в столице нашей страны [11]. На сегодняшний день расшифрована структура почти 6000 олигопептидов, выделенных из представителей всех царств живого. Так же как и крупные белки, количество олигопептидов с заданным числом аминокислотных остатков можно изобразить графически (рис. 10). Судя по графику, чаще всего встречаются олигопептиды, содержащие примерно 8–10 аминокислотных остатков. Среди них в основном содержатся молекулы, которые участвуют в регуляции нервной системы, и поэтому называются нейропептидами. Очевидно, что самые быстрые процессы в живом организме осуществляются с участием нервной системы, поэтому пептидные регуляторы должны быть мобильными и следовательно небольшими. Однако, следует отметить, что, ввиду огромного структурного и функционального разнообразия как белков, так и пептидов, для них до сих пор не создано строгой классификации.
Таким образом, в данном случае задачами биоинформатики являются накопление информации о физико-химических и биологических свойствах белков, анализ этой информации, каталогизация и подготовка информационной базы и вычислительных средств для выявления механизмов их функционирования.
Функциональная протеомика
Наличие в организме того или иного белка дает основание предполагать, что он обладает (или обладал) определенной функцией, а весь протеом служит для того, чтобы осуществлялась полноценная жизнедеятельность всего организма. Функциональная протеомика занимается определением функциональных свойств протеома, и решаемые ею задачи существенно сложнее, чем, например, определение белково-пептидных структур.
Очевидно, что функционирование протеома осуществляется в многокомпонентной среде, в которой присутствует множество молекул других химических классов – сахаров, липидов, простагландинов, различных ионов и многих других, включая молекулы воды. Не исключено, что через некоторое время появятся такие термины, как «сахаром», «липидом» и им подобные. Белковые молекулы взаимодействуют с окружающими их другими или такими же, как и они, структурами, что в конечном итоге приводит к возникновению функциональных реакций сначала на молекулярном уровне, а затем и на макроскопическом. Уже известно множество таких процессов, в том числе с участием белков. Среди них взаимодействие фермента с субстратом, антигена с антителом, пептидов с рецепторами, токсинов с ионными каналами и т.д. (рецепторы и ионные каналы также являются белковыми образованиями). Для выявления механизмов этих процессов проводятся как экспериментальные исследования индивидуальных участников взаимодействия, так и системные исследования средствами биоинформатики. Рассмотрим несколько примеров таких системных подходов.
На рис. 11 показаны представители протеома (в данном случае пептидома) человека – различные гастрины и холецистокинины, которые локализованы в желудочно-кишечном тракте (при написании аминокислотных последовательностей использован стандартный однобуквенный код, расшифровка которого была дана нами ранее [7]). Функциональными частями молекул этих пептидов являются очень схожие правые области. Однако пептиды обладают прямо противоположными поведенческими свойствами: гастрины вызывают у человека ощущение голода, а холецистокинины – сытости. По-видимому, данное различие обусловлено тем, что в первичной последовательности холецистокининов положение остатка тирозина Y сдвинуто на один шаг по сравнению с гастринами. На том же рисунке приведена первичная структура пептида ционина, полученного из представителя простейших хордовых Ciona intestinalis (рис. 12). Его структура гомологична и гастринам, и холецистокининам и характеризуется двумя остатками тирозина, находящимися в тех же положениях, что и у обоих указанных пептидов. К сожалению, функциональные свойства его не изучены. А при должном экспериментальном исследовании можно было бы ответить на вопрос, какова роль химической структуры в целом и остатков тирозина в частности при проявлении противоположных физиологических эффектов.

Рис. 11. Первичные структуры представителей пептидома человека в сравнении со структурой одного из пептидов оболочечника

Рис. 12. Оболочечник Ciona intestinalis, обитающий в Северном море
Другой пример: на рис. 13 приведены аминокислотные последовательности очень похожих молекул, которые также объединены в структурно-гомологичное семейство. Эти молекулы обнаружены у весьма эволюционно далеких живых организмов – от насекомых до млекопитающих. В первой строке дана первичная структура брадикинина, содержащего 9 аминокислотных остатков и встречающегося у многих высших организмов, в том числе и у человека. В течение многих лет химики синтезировали различные неприродные аналоги этой молекулы, чтобы ответить на вопрос, какой ее участок ответственен за взаимодействие с рецептором. Около 30 лет назад были даже синтезированы все возможные фрагменты брадикинина – 8 дипептидов, 7 трипептидов и т.д. (всего возможны 36 фрагментов), величину активности которых затем испытывали в одном и том же биологическом тесте. Результат оказался тривиальным: выяснилось, что максимальную активность проявляет лишь вся молекула целиком, а каждый фрагмент по отдельности обладает либо следовой активностью, либо нулевой. Эту трудоемкую работу не пришлось бы делать, если бы в то время были известны другие брадикинины, приведенные на рис. 13, и средствами биоинформатики они были бы выделены из глобального протеома. Представленное структурно-гомологичное семейство наглядно демонстрирует, что у всех молекул есть область, которая в результате биологической эволюции практически не изменялась (квазиконсервативная область), и она представляет собой молекулу брадикинина высших живых организмов, отобранную как наиболее совершенную в результате эволюционного процесса. Данный пример демонстрирует, что протеомика вместе с биоинформатикой позволяет быстро (и дешево) решать принципиальные научные проблемы.

Рис. 13. Первичные структуры природных пептидов брадикининов, полученных из разных живых организмов. Жирным шрифтом указаны квазиконсервативные области

Рис. 14. Первичные структуры структурно-гомологичного семейства эндотелинов / токсинов
И, наконец, третий пример – структурно-гомологичное семейство эндотелинов млекопитающих и токсинов змей (рис. 14). Несмотря на поразительное сходство структур, их функциональные свойства разительно отличаются друг от друга: одни являются очень полезными регуляторами сосудистого сокращения, а другие – смертельно опасны для жизни. В данном случае мы сталкиваемся с ситуацией, когда первичная структура не несет достаточной информации, способной объяснить причину различия функций, и необходимо более детальное рассмотрение пространственной (третичной) структуры. На рис. 15 и 16 показаны пространственные структуры двух представителей этого семейства – эндотелина-1 и сарафотоксина 6b, полученные с помощью ЯМР-спектроскопии. На рисунках они повернуты так, чтобы достичь максимальной пространственной гомологии. Но полной гомологии не удается получить ни при каком повороте. Следовательно, несмотря на большое сходство первичных структур, взаимодействие их осуществляется с разными рецепторными структурами, а потому и приводит к разным физиологическим эффектам.
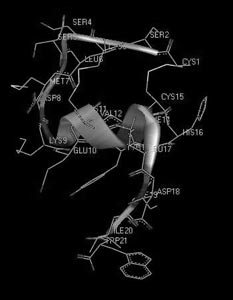
Рис. 15. Пространственная структура сосудосокращающего пептида эндотелина-1 человека
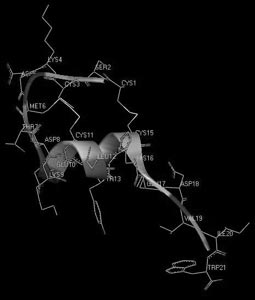
Рис. 16. Пространственная структура сарафотоксина 6b израильской змеи Atractaspis engaddesis
Конечно столь частными примерами невозможно полностью охарактеризовать многообразие функциональной протеомики. Создание представлений об огромной сети взаимодействий белковых и других молекул в организме требует огромного труда и применения всех средств современной биоинформатики. По существу, создание таких представлений еще только начинается. Однако есть основание полагать, что с каждым годом наши познания в этой области будут быстро расти.
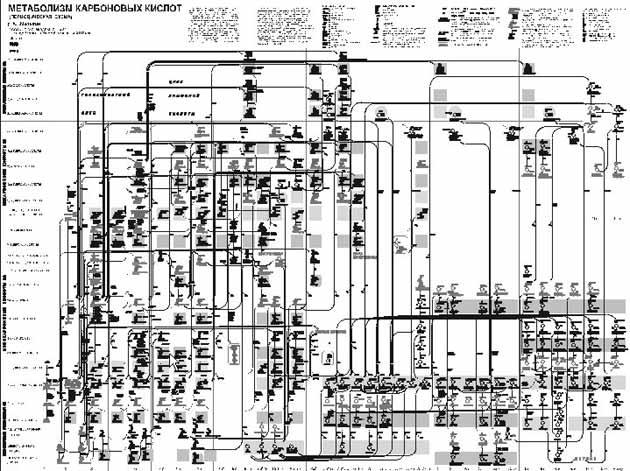
Рис. 17. Общие контуры карты метаболизма карбоновых кислот [12]
Одним из первых успехов на этом пути является создание карты метаболизма карбоновых кислот в Институте биохимии им. А.Н. Баха Российской академии наук (рис. 17). Эта карта представляет собой сеть реакций с регулярным периодическим строением. Такой подход оказался успешным ввиду того, что функционально аналогичные метаболиты претерпевают сходные биохимические превращения, образуя функционально аналогичные производные. В карте по вертикали расположены области, содержащие соединения с одинаковым числом атомов углерода (от 1 до 10), а горизонтальные ряды представляют собой ряды функционально аналогичных метаболитов. Химические структуры на карте соединены многочисленными стрелками с указанием, какие ферменты (белки) участвуют в соответствующих химических превращениях. Не правда ли, такой подход напоминает периодическую систему химических элементов Д.И. Менделеева? И так же, как и менделеевская система, данная карта обладает прогностической силой. С ее помощью был предсказан целый ряд новых ферментов, которые впоследствии были обнаружены экспериментально.
Подобные схемы могут быть распространены и на другие метаболические процессы (например, углеводов, аминокислот и т.д.), а также использованы для поиска новых метаболитов биохимических реакций.
Таким образом, функциональная протеомика занимается изучением сложных взаимосвязей структуры и функций протеома.
Практическая протеомика
Итак, главной задачей протеомики является выявление механизма взаимодействия огромного числа белков и пептидов в одном организме. Какова же практическая значимость этой грандиозной и дорогостоящей работы? Очевидно, что в первую очередь в результатах такой работы заинтересованы фармакологи и медики, поскольку очень часто прослеживается тесная связь между изменениями в белковом составе и болезненным состоянием человека. Поэтому новые данные в протеомике будут использоваться (и уже используются) для быстрой разработки новых лекарственных средств и новейших методов лечения болезней, с которыми медицина боролась веками. На сегодняшний день 95% всех фармакологических средств воздействуют на белки. Протеомика со своим системным подходом может помочь идентифицировать и оценить важность появления новых белков гораздо эффективнее, что, в свою очередь, ускорит разработку новых диагностических тестов и терапевтических средств.
Первое практическое применение протеомных исследований состоялось задолго до появления термина «протеомика», еще в начале XX в., когда была обнаружена роль инсулина в развитии такого тяжелого заболевания, как диабет. Создание инсулиновых препаратов спасло жизнь миллионам людей.
В настоящее же время протеомика, вместе с геномикой и биоинформатикой, ориентирована на создание новых лекарственных препаратов (рис. 18), в которых молекулярными мишенями будут служить те или иные белки [13]. Процесс нахождения новых мишеней для действия лекарств решается с помощью биоинформатики, причем объектом анализа является геном. Однако после анализа генома необходимо получить доказательства того, что данный белок интенсивно экспрессируется и находится в клетке в рабочем состоянии. Эту задачу решает протеомика. Таким образом выявляется молекулярная генетическая мишень для лекарства.
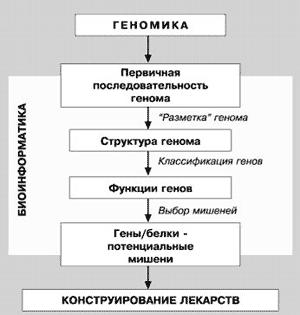
Рис. 18. Взаимосвязь геномики, протеомики и биоинформатики при решении проблемы конструирования новых лекарственных средств
Следует отметить, что протеомика может и сама по себе решать проблему нахождения мишени. Если получить протеомные карты (подобные тем, что представлены на рис. 3 или 4) нормальных и патологических тканей, то по различиям в них можно установить, какие белки важны для развития того или иного патологического состояния, и выбрать их в качестве мишеней или использовать эти знания для диагностики. Можно предположить, что в будущем к обычному анализу крови добавится создание протеомных карт крови. Для этого в поликлиниках необходимо будет использовать специальное оборудование, с помощью которого у пациентов периодически будут брать кровь. При возникновении болезненного состояния протеомную карту больного человека нужно будет всего лишь сравнить с его же протеомной картой, но составленной в то время, когда он был здоров, и можно будет выявить произошедшие изменения в белковом составе крови и определить причину заболевания. Подобное сравнение протеомов опухолевых и нормальных клеток, клеток до и после воздействия определенных факторов (например, физических или химических), использование биологических жидкостей в диагностических целях – все это представляет огромный интерес и открывает совершенно новые перспективы для медицины, ветеринарии, фармакологии, пищевой промышленности и других прикладных областей. Впереди предстоит огромная и интересная работа.
Список литературы
1. Sanger F., Air G.M., Barrell B.G., Brown N.L. et al. Nucliotide sequence of bacteriophage phi X-174 DNA.//Nature. 1977. V. 265, № 5596. P. 687–695.
2. Fleischmann R.D., Adams M.D., White O. et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd.//Science. 1995. V. 269, № 5223. P. 496–512.
3. Nature. 2001. 409, № 6822 (большая часть выпуска журнала посвящена расшифровке генома человека).
4. Ferguson-Smith A.C., Ruddle F.H. The genomics of human homeobox-containing loci.//Pathol. Immunopathol. Res. 1988. V. 7, № 1–2. P. 119–126.
5. Franklin J. Bioinformatics changing the face of information.//Ann. NY Acad. Sci. 1993. V. 700. P. 145–152.
6. Wasinger V.C., Cordwell S.J., Cerpa-Poljak A. et al. Progress with gene-product mapping of the Mollicutes: Mycoplasma genitalium.//Electrophoresis. 1995. V. 16, № 7. P. 1090–1094.
7. Замятнин А.А. Блистающий мир белков и пептидов.//Биология. 2002. № 25–26. P. 8–13.
8. Gorg A., Weiss W., Dunn M.J. Current two-dimensional electrophoresis technology for proteomics.//Proteomics. 2004. V. 4, № 12. P. 3665–3685.
9. Ramstrom M., Bergquist J. Miniaturized proteomics and peptidomics using capillary liquid separation and high resolution mass spectrometry.//FEBS Lett. 2004. V. 567, № 1. P. 92–95.
10. http://au.expasy.org/sprot/
11. http://erop.inbi.ras.ru/
12. Малыгин А.Г. Метаболизм карбоновых кислот (периодическая схема). – М.: «Международная программа образования», 1999.
13. Арчаков А.И. Что за геномикой? – Протеомика.//Вопр. мед. химии. 2000. Т. 46, № 4. С. 335–343.