Суммарные показатели качества сточных вод
СОДЕРЖАНИЕ: Определение общего содержания серы в сточных водах. Анализ вод методом Кьельдаля. Ход и условия проведения определения запаха и цвета воды. Тяжелые металлы, суммарное определение, сущность метода. Общее содержание азота и азоторганических веществ.СУММАРНЫЕ ПОКАЗАТЕЛИ КАЧЕСТВА ВОД
ЦВЕТ ВОДЫ
Цвет воды рекомендуется определять измерением ее оптической плотности на спектрофотометре при различных длинах волн проходящего света.
При определении цветности пробы не консервируют. Определение проводят через 2 ч после отбора пробы.
Исследуемую воду предварительно профильтровывают, отбрасывая первые порции фильтрата. Оптическую плотность измеряют при толщине слоя 10 см, вторую кювету прибора заполняют дистиллированной водой. Длина волны света, максимально поглощаемого водой, является характеристикой ее цвета. Если на полученной кривой имеется несколько пиков, то соответствующие им длины волн должны быть отмечены.
Следует учитывать, что видимый цвет раствора всегда является дополнительным к цвету поглощаемого излучения.
Значение оптической плотности исследуемой воды при длине волны, близкой к максимуму поглощения, является мерой интенсивности ее окраски.
Спектрофотометр может быть заменен фотометром (типа прибора Пульфриха) или электрофотоколориметром при наличии достаточного числа светофильтров, пропускающих узкие полосы спектра света. В отсутствие приборов цвет сточной воды приходится определять визуальным наблюдением: светло-желтый, пурпурно-красный и т. д.
Поскольку по правилам спуска сточных вод в водоем требуется, чтобы вода в водоеме после смешения ее со сточной водой не имела видимой окраски при толщине слоя 10 см, практическое значение имеет определение степени разбавления сточной водой, при котором цвет ее при указанной толщине слоя перестает различаться. Для этого помещают на лист бумаги три цилиндра (диаметром 20—25 мм) из бесцветного стекла. В первый наливают исследуемую сточную воду (высота слоя 10 см), в третий — такое же количество дистиллированной воды; во второй цилиндр наливают в таком же объеме разбавленную сточную воду, увеличивая каждый раз степень разбавления (1:1; 1:2; 1 :3 и т. д.), пока при просматривании сверху через воду во втором и третьем цилиндрах бумага не будет выглядеть одинаково белой. Нахождение того разбавления, которое требуется для исчезновения окраски, устраняет необходимость определять количественное содержание в воде всех тех веществ, для которых установлена ПДК по цветности.
ЗАПАХ ВОДЫ
Качественное определение запаха проводят как при комнатной температуре, так и при нагревании до 60 °С в колбе, покрытой часовым стеклом. Результат этого определения выражают описательно: хлорный — запах свободного хлора, землистый — запах влажной почвы, фенольный, запах нефти, аптечный, сероводородный, навозный, затхлый, запах гнилого сена, гнилостный и т. п.
Для количественного определения запаха находят так называемое пороговое число, выражающее во сколько раз надо разбавить анализируемую воду чистой, не имеющей запаха, водой, чтобы запах пробы перестал ощущаться. Для разбавления следует применять водопроводную воду, предварительно пропущенную через колонку с активным углем. Дистиллированную воду применять не следует, так как она часто имеет своеобразный запах.
В тех случаях, когда запах сточной воды вызван присутствием в ней веществ, имеющих кислотные или основные свойства, за-1 пах надо определять при том значении рН, при котором он наиболее ощутим. Это значение рН находят экспериментально, приготовив ряд буферных растворов (рН = 5; 6; 7; 8; 9) и прибавив в каждый раствор одинаковое (не слишком большое) количество анализируемой пробы. Последующее определение «порогового числа» анализируемой воды производят, применяя разбавляющую воду, доведенную соответствующим буферным раствором до этого критического значения рН.
Условия проведения определения. Все определения надо проводить в комнате, куда не могут проникнуть какие-либо запахи. Аналитики не должны перед проведением испытаний курить, принимать пищу, пользоваться духами, одеколоном, лосьонами и т. п Они не должны в это время болеть насморком или страдать какой-либо аллергией. Поскольку способность ощущать очень слабые запахи у людей различна, это определение должно сопровождаться одновременно несколькими аналитиками — не менее чем тремя. Исключительной сенситивности здесь не требуется, но людей с притуплённым обонянием допускать к таким определения нельзя. Рекомендуется проводить предварительную проверку: из4 бранной группе аналитиков дать определять пороговое число раствора какого-нибудь чистого пахучего вещества. Для этой цели предложены бутанол и м-крезол. Сильно пахнущие сточные воды надо предварительно во много раз разбавить и это разбавление учесть при расчете результата определения. Определение запаха нельзя проводить, дольше 1 ч, потому что обоняние быстро притупляется.
Если анализируемая вода подвергалась хлорированию, то в таких случаях рекомендуется проводить два определения запаха: одно— без каких-либо изменений пробы, второе — после ее дехлорирования добавлением эквивалентного количества аскорбиновой кислоты или тиосульфата.
Ход определения. Пороговое число находят следующим способом. В конические колбы вместимостью 500 мл, снабженные стеклянными пробками, наливают немного разбавляющей воды, затем вводят 2; 5; 10; 50; 150 мл анализируемой воды, разбавляют содержимое каждой колбы до 200 мл разбавляющей водой и перемешивают, закрыв, стеклянной пробкой. Еще в одну колбу вместимостью 500 мл наливают 200 мл разбавляющей воды. Вынув пробки, попеременно подносят к носу то одну из колб с пробой, то колбу с чистой водой. Часто предпочитают при этом пользоваться специальным приспособлением — стеклянной трубкой диаметром 1,8 см, длиной 10—30 см, расширенной у верхнего конца так, чтобы она охватывала обе ноздри. Трубку эту надо предварительно промыть разбавляющей водой.
Определив таким способом приближенно наибольшее разбавление, при котором запах будет все же ощущаться, исходя и» этого результата, проводят более точное измерение, приготовив вторую серию разбавлений пробы с меньшими разностями концентраций между соседними растворами. Наименьшее из разбавлений, при котором запах исчезает, и есть пороговое число.
Определение проводят также при 60 °С, для чего колбы с растворами погружают в соответственно нагретую водяную баню. Колебания в температуре не должны превышать 1 °С.
Если известно, что анализируемая вода содержит только одно какое-либо пахнущее вещество, то описанным выше методом можно определить его концентрацию в пробе. Для этого определяют пороговое число, запах пробы и пороговое число запаха стандартного раствора этого пахучего вещества известной концентрации. Тогда концентрация этого вещества (с*) в пробе будет равна
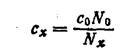
Определение порогового числа избавляет от необходимости определять количественное содержание в воде всех тех веществ, для которых ПДК установи лена по запаху. (Исключением являются лишь фенолы, для которых ПДК установлены не по запаху самих фенолов, а по запаху продуктов их хлорирования).
ТЯЖЕЛЫЕ МЕТАЛЛЫ, СУММАРНОЕ ОПРЕДЕЛЕНИЕ
Сущность метода. Тяжелые металлы — свинец, медь, кадмий, цинк, никель — извлекаются из раствора в виде их дитизонатн комплексов, избыток дитизона удаляют, полученную смесь дитзонатов обрабатывают солью ртути(II). Поскольку ртуть образу с дитизоном наиболее устойчивое комплексное соединение, тизонаты всех перечисленных металлов превращаются в дитизонат ртути Hg(HDz)2 . Измеряют оптическую плотность его раствора при X = 485 нм (е = 7 • 104 ).
Результат определения выражают в мэкв/л.
Мешающие вещества. Поскольку тяжелые металлы в анализируемой пробе могут быть в виде комплексных соединений с ионами или анионами различных органических кислот, необходимо предварительное разрушение этих комплексов одним методов. Если присутствуют бихромат ионы, их восстанавливают добавлением гидразина или какого-нибудь другого восстановителя. Ионы трехвалентного железа связывают прибавлением раствора тартрата натрия.
Реактивы
Вода дистиллированная (встряхивают с малыми порциями раствора дитизона до тех пор, пока цвет прибавленной порции не перестанет изменяться) промытая несколькими порциями чистого CCI4.
Дитизон, раствор в тетрахлориде углерода. Тартрат натрия, 5%-ный раствор, очищенный раствором дитизона.
Аммиак, концентрированный раствор.
Промывной раствор, 1 мл концентрированного раствора аммиака разбавляют до 1 л дистиллированной водой.
Тетрахлорид углерода чда.
Нитрат ртути, растворяют 10 мг соли в 1 л 2 н. уксусной кислоты.
Стандартный раствор соли тяжёлого металла. Основной раствор, содержащий 1 микрограмм-эквивалент (0,001 миллиграмм-эквивалент) металла в 1 и готовят растворением 0,1542 г нитрата кадмия Cd(N03 )s-4HjO или 0,1438г сульфата цинка (ZnS04-7H2 0) или 0,1656 г нитрата свинца в дистиллирова:ной воде и разбавлением до 1 л.
Рабочий раствор готовят разбавлением 10 мл основного раствора дистилированной водой до 100 мл; в 1 мл полученного раствора содержится 0,1 мкг грамм-эквивалента (10-4 миллиграмм-эквивалента) тяжелого металла.
Гидрохлорид гидроксиламина, 5%-ный раствор. ХОД определения. Помещают 500 мл пробы (или меньший объем разбавленный до 500 мл), выпаривают в фарфоровую чашку малого объема и обрабатывают азотной кислотой и пероксид водорода. Остаток растворяют в 50 мл очищенной дитизоном дистиллированной воды и фильтруют через быстро фильтрующий фильтр в коническую колбу. Прибавляя 10 мл 5%-ного раствора гидрохлорида гидроксиламина и нагревают 10 мин при 80—90 °С. Затем охлаждают, переносят раствор в делительную воронку, прибавляют 10 мл 5%-ного раствор тартрата натрия и добавлением около 1 мл концентрированного раствора аммиака доводят рН раствора до 8—8,5 по универсальной индикаторной бумаге.
Проводят экстракцию, внося раствор дитизона порциями по 10 и 5 мл и сильно встряхивая 2 мин. Растворы дитизона объединяют. Добавление порций дитизона прекращают, когда окраска их после встряхивания перестанет изменяться. После этого еще один раз экстрагируют 5 мл тетрахлорида углерода. Присоединяют полученный экстракт к дитизоновым растворам, переносят в делительную воронку и обрабатывают 2—3 раза промывным раствором, порциями по 10 мл. К отмытым экстрактам приливают 50 мл 0,01%-ного раствора нитрата ртути, встряхивают 2, мин и после расслоения сливают слой тетрахлорида углерода, содержащий оранжево-желтый дитизонат ртути, в мерную колбу вместимостью 50 мл, доводят до метки тетрахлоридом углерода и перемешивают.
Измеряют оптическую плотность при % = 485 нм по отношению к раствору, полученному в холостом опыте, в котором 500 мл дистиллированной воды, очищенной дитизоном, проводят через всё стадии хода определения.
Предельно допустимые концентрации (ПДК) тяжёлых металлов, выраженные в мэкв/л располагаются в следующем ряду по возрастанию: кадмий (1,8-10-4 ), свинец (9,7-10~4 ), медь (3,1-10-3 ), никель (3,4-10-3 ), цинк (3,1 • 10-2 ). Если найденное суммарное содержание тяжелых металлов превосходит первое из этих значений (1,8•10-4 ), приступают к определению содержания отдельных металлов, начиная с кадмия, и вычитая из суммы найденный результат определения до тех пор, пока разность не окажется ниже ПДК следующего по порядку металла. Очевидно, что если заранее известно отсутствие в анализируемой воде какого-либо из указанных металлов, то его определение исключают.
УГЛЕРОД ОРГАНИЧЕСКИЙ
Определение органического углерода — одно из основных определений при выполнении органического элементного анализа. Это «классическое» определение осуществляют известными точными методами, в которых органическое вещество сжигают «сухим» способом в токе кислорода (после выпаривания пробы) или «мокрым» способом — обработкой пробы окислителем (К2 Сг2 О7 , СгО3 или K2 S2 O8 ) в растворе H2SO4 в присутствии катализатора, Ag2 S04. Образующийся в обоих случаях СО2 поглощают КОН или Ва(ОН)2 и заканчивают определение гравиметрическим или титриметрическим методом. В последнее время конечное определение СО2 стали проводить ИК-спектрометрическим методом более простым в выполнении и легко автоматизируемым.
Сравнительно недавно появились приборы, в которых анализируют очень малые порции сточных или природных вод (всего лишь 20—40 мкл), и все определение занимает несколько (3— 5) минут. Выпуск таких приборов подготовляется нашей промышленностью, и эти приборы, надо полагать, поступят в продажу, в ближайшее время. В таком приборе малую порцию сточной воды быстро выпаривают и сжигают в токе воздуха при 950 °С в присутствии катализатора — оксида кобальта, нанесенного на асбест. Количество образующегося С02 измеряют в ИК-анализаторе по длине (высоте получаемого очень узкого пика.
В тот же прибор одновременно вводят вторую порцию пробы, которую в другой части прибора выпаривают и нагревают при 150 °С в трубке, заполненной кусочками кварцевого стекла, смоченными 85%-ной фосфорной кислотой. В этих условиях выделяется только свободный и карбонатный диоксид углерода. Количество СО2 измеряют также в ИК-анализаторе. Разность между длинами (высотами) пиков, полученными в той и другой части прибора, выражает количество СО2 , образовавшегося при сжигании органических веществ пробы.
Метод требует предварительного фильтрования пробы и, следовательно, показывает содержание органического углерода только в жидкой фазе пробы. Если по какой-либо причине требуется определение суммарного содержания органического углерода в обеих фазах, твердой и жидкой, то для анализа приходится брать относительно большой объем хорошо гомогенизированной пробы.
ОБЩЕЕ СОДЕРЖАНИЕ АЗОТА («ОБЩИЙ АЗОТ») И АЗОТ ОРГАНИЧЕСКИХ ВЕЩЕСТВ («ОРГАНИЧЕСКИЙ АЗОТ»)
Сущность метода. При нагревании органических веществ со смесью концентрированной серной кислоты и сульфата калия (т. кип. 315—370 °С) в присутствии катализатора — соли меди или ртути — Происходит разложение этих веществ с образованием сульфата аммония (метод Кьельдаля). Раствор подщелачивают, отгоняют аммиак и определяют его в отгоне. Так находят суммарное содержание азота органических соединений и азота, который первоначально был в пробе в виде аммонийной соли. Вычитая из полученного результата отдельно найденное содержание азота аммонийного, получают содержание в пробе азота органических веществ «органический азот».
Нитраты и нитриты в ходе этого процесса разлагаются с выделением и улетучиванием образующихся оксидов азота. Их определяют отдельно соответствующими методами, присоединяют полученные результаты в пересчете на азот к результату определения органического азота и получают содержание общего азота».
Следует указать на то, что ряд органических соединений не превращается (или лишь частично превращается) в аммонийную соль в ходе этого процесса: гетероциклические соединения, содержащие азота ядре, азиды, азины, азосоединения, нитрилы, нитро-и нитрозосоединения, оксимы, семикарбазоны. Это делает условными сами понятия: «общий азот», «общий, органический азот», поэтому термины поставлены здесь в кавычки. Еще лучше указывать на метод конечного определения: «Общий азот во Кьельдалю», «Органический азот по Кьельдалю».
Реактивы Дистиллированная вода, не содержащая аммонийных солей и аммиака.
Обычную дистиллированную воду подкисляют, прибавляют к ней перманганат калия и перегоняют. Эту операцию повторяют еще раз. Как перегонку воды, так и само определение азотсодержащих веществ надо проводить в комнате, где нет аммиака в воздухе. Очистить дистиллированную воду от аммиака и аммонийных солей можно также, пропуская ее через слой катионита. При прохождении через катионит ионы аммония обмениваются на ионы водорода.
Серная кислота, пл. 1,84 г/см8 , не содержащая оксидов азота (хч или чда), 0,02 н. стандартный раствор; 1 мл этой кислоты соответствует 0,28 мг азота.
Едкий натр, 50%-ный раствор, t Сульфат меди CuS04-5H2 0, 10%-ный раствор.
Борная кислота. Растворяют в воде, не содержащей аммиака и солей аммония, 40 г борной кислоты и разбавляют раствор такой же водой до 1 л.
Бромфеноловый синий (растворяют в 3,0 мл 0,05 н. раствора едкого натра 0,1 г бромфенолового синего и разбавляют раствор до 100 мл дистиллированной водой) или метиловый красный (растворяют в 7,4 мл 0,05 н. раствора едкого натра 0,1 г метилового красного и разбавляют раствор до 100 мл дистиллированной водой).
Фенолфталеин, 1 %-ный спиртовый раствор.
Сульфат калия или сульфат натрия, безводный. Сульфид натрия Na2 S-9H2 0, 4%-ный раствор.
Пемза. Кусочки пемзы кипятят несколько раз в дистиллированной воде, сливая воду после каждого кипячения, и высушивают.
Ход определения. Берут такой объем анализируемой сточной воды, чтобы в ней содержалось 2—6 мг азота (в виде органических соединений и солей аммония), переносят в колбу Кьельдаля, прибавляют 10 мл серной кислоты (пл. 1,84 г/см3 ), 5 г сульфата калия или сульфата натрия, 1 мл раствора сульфата меди и всыпают несколько кусочков пемзы[1] . Содержимое колбы кипятят; сначала удаляется вода, потом напитается разложение органических веществ и жидкость в колбе становится темной. Кипячение продолжают до тех пор, пока раствор в колбе не станет вполне прозрачным и бесцветным или слабо-зеленоватым. Охладив колбу, переносят жидкость (вместе с кусочком пемзы) в перегонную колбу прибора для отгонки аммиака. Стенки первой колбы обмывают примерно 250 мл дистиллированной воды, не содержащей аммиака.
Прибавляют 2,5 мл раствора сульфида натрия, 3—5 капель раствора фенолфталеина и затем осторожно по стенке колбы, так, чтобы жидкости не смешивались, наливают 50 мл раствора едкого натра. Сейчас же включают колбу в собранную установку для отгонки (в приемник наливают 50 мл раствора борной кислоты), осторожно вращая, смешивают в ней оба слоя жидкости (жидкость должна окраситься в красный цвет) и начинают нагревание. Конец трубки холодильника должен быть погружен в раствор борной кислоты, находящийся в приемнике.
Отгонку заканчивают, когда в перегонной колбе останется меньше первоначального объема жидкости. Затем отъединяют приемник, наливают в него несколько капель раствора бромфенолового синего или метилового красного и титруют 0,02 н. раствором серной кислоты до изменения окраски индикатора, пользуясь «свидетелем», для приготовления которого к такому же объему дистиллированной воды, освобожденной кипячением от СО2 , прибавляют те же количества раствора борной кислоты и индикатора, какие были введены в приемник при анализе пробы. Проводят холостой опыт со всеми примененными в анализе реактивами.
Расчет. Общее содержание азота (х) в мг/л вычисляют по формуле
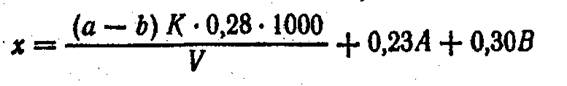
Взамен раствора борной кислоты можно в приемник налить 0,02 н. раствор серной кислоты и оттитровать избыток этой кислоты 0,02 н. раствором едкой щелочи. Содержание азота органических соединений (у) в мг/л находят во формуле
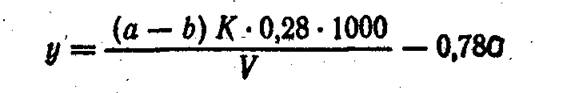
АЗОТСОДЕРЖАЩИЕ ВЕЩЕСТВА, ПОКАЗЫВАЮЩИЕ ПОЛОЖИТЕЛЬНУЮ РЕАКЦИЮ НА НИНГИДРИН (NPS)
Сущность метода. Положительную реакцию на нингидрин (образование с этим реактивом соединения, окрашивающего раствор в фиолетовый цвет) показывают в основном алифатические азотсодержащие вещества (амины и имины), аминосахара, амино- и иминокислоты, пептиды, содержащие в молекуле менее 10 аминокислотных группу и др. Такие вещества, |как п-фенилендиамин, n-аминофенол, аминотиолы, дают эту реакцию лишь в очень слабой мере (примерно в 10 раз слабее, чем основные вещества, перечисленные выше). Совсем слабо реагируют полипептиды и некоторые соединения, не содержащие азот (оксальдегиды, оксикетоны, катокислоты).
Таким образом, «проба на нингидрин» (NPS), определяя некоторую часть из всех азотсодержащих веществ в анализируемой воде, практически полностью определяемых методом Кьельдаля позволяет разделить эти вещества на две группы: дающие положительную NPS-реакцию и не дающие этой реакции.
Нередко при анализе вод такое разделение оказывается полезным. Кроме того, следует учесть, что проба на NPS-вещества очень проста в выполнении, ее можно провести значительно быстрее анализа по Кьельдалю, поэтому нередко бывает достаточно только этой пробы.
Как и при анализе по Кьельдалю, азот NH3 и NHt включается в результате определения, его надо определять отдельно и вычитать из полученного результата. Результат проведения пробы выражают символом NN ps.
В ходе реакции молекулы нингидрина и продукта его восстановления аминосоединения или аммиака — соединяются, образуя
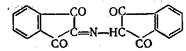
вещество, которое затем превращается в фиолетовый Руэманна:
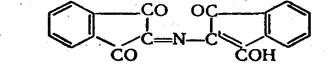
Казалось бы, поэтому, что количество окрашенного продукта и интенсивность получаемой окраски не должны зависеть от тогокакие именно азотсодержащие вещества вступили в реакцию с нингидрином. Однако одновременно с основной реакцией проходят и некоторые побочные реакции, вследствие которых цвет раствора может несколько различаться по оттенку и коэффициент поглощения колебаться от 1,8-104 до 3,3 • 104 . Оптическую плотность измеряют при = 570 нм и в качестве стандарта для построения калибровочного графика всегда берут хлорид аммония.
Мешающие вещества. В сточных водах могут содержаться вещества, ингибирующие реакцию с нингидрином. Устранить их влияние разбавлением пробы невозможно. При подозрении на возможность их присутствия следует применять метод стандартной добавки и, если окажется необходимым, вводить соответствующую поправку. Если проба содержит тяжелые металлы (медь, кадмий, железо) в концентрациях, превышающих 5 мг/л, надо ввести соотвественно большее количество цианида.
Реактивы
Нингидрин, 3%-ный раствор. Быстро отвешивают 1,50 г нингидрина и немедленно растворяют в 50 мл смеси (1:1) этилового и изопропилового спиртов. Поскольку раствор устойчив не более трех суток, рекомендуется приготовлять небольшие порции его перед применением. Хранить раствор надо в плотно закупоренном сосуде в холодильнике.
Дистиллированная вода, не содержащая аммиак и аммонийные соли .
Буферный уксусно-ацетатный раствор. Растворяют 270 г ацетата натрия в 250 мл безаммиачной воды, прибавляют 50 мл ледяной уксусной кислоты и разбавляют безаммиачной водой до 500 мл. Раствор можно хранить около 6 месяцев.
Цианид калия, 0,001 М раствор. Приготовляют из более концентрированного перед применением.
Этиловый (96%-ный) и изопропиловый спирты.
Стандартный раствор хлорида аммония. Основной: растворяют 0,7638 р хлорида аммония хч, предварительно высушенного в эксикаторе, в безаммиачной воде и разбавляют такой же водой до 1 л. Рабочий: разбавляют 25 мл основного раствор безаммиачной водой до 1 л. Приготовляют перед применением. 1 мл основного раствора содержит 200 мкг азота, 1 мл рабочего раствора — 5 мкг азота.
Ход определения. Целесообразно обрабатывать одновременно серию из 7—10 проб. К 20 мл каждой пробы, которые должны содержать 0,5—10 мкг азота каждая, помещенным в мерные колбы вместимостью 25 мл или в градуированные пробирки, снабженные притертыми стеклянными пробками, приливают по 0,5 мл буферного раствора. При анализе более концентрированных растворов берут меньшие объемы пробы и разбавляют безаммиачной водой до 20 мл; рН растворов должен быть в пределах от 3 до 9. В каждый сосуд вливают по 1 мл раствора нингидрина и по 0,1 мл раствора цианида калия, закрывают пришлифованными пробками и перемешивают. Затем, прежде чем нагреть пробы вынимают стеклянные пробки, кладут их на чистую подставку и оберегают от пыли. Взамен пробок каждый сосуд обертывают у, горлышка алюминиевой фольгой (3 см X 3 см) и ставят все сосуды в кипящую водяную баню, заполненную безаммиачной водой, точно на 15 мин. Вынув сосуды из ванны, немедленно в еще горячие растворы вливают изопропиловый спирт примерно до объема 23—24 мл, вставляют взамен фольги притертые пробки и перемешивают. Дают сосудам охладиться до комнатной температуры (примерно через 30 мин), доливают в каждую пробу изопропиловый спирт до метки 25,0 мл, перемешивают и в течение 1 ч измеряют оптическую плотность в кювете с толщиной слоя 1—3 см по отношению к чистой безаммиачной воде при 2= 570 нм. Для каждой серии проб проводят холостое определение, и результат его учитывают при расчете.
Для построения калибровочного графика отбирают порции рабочего стандартного раствора хлорида аммония, содержащие от 1 до 10 мкг азота, разбавляют каждую порцию до 20 мл и проводят определение так же, как анализ проб.
ОБЩЕЕ СОДЕРЖАНИЕ СЕРЫ («ОБЩАЯ СЕРА»)
В сточных водах могут присутствовать различные соединения содержащие серу: неорганические — сульфаты, сульфиты, сульфиды, тиосульфаты, роданиды, свободная сера и т. п., органические — белковые соединения, органические сульфиды, дисульфиды, меркаптаны, различные сульфосоединения, поверхностно-активные моющие вещества и многие другие. Серу в этих соединениях объединяют названием «общая сера». Определив ее содержание в сточной воде, можно затем отдельно определить серу сульфатов, сульфитов, сероводорода, роданидов, меркаптанов и т. д., уменьшая таким образом величину рассчитываемого по разности остатка, который записывают в таблицу результатов под названием «сера других химических соединений».
Для определения общего содержания серы прежде всего окисляют все содержащие этот элемент вещества; сера при этом превращается в сульфат-ионы, затем определяют содержание последних. Окисление, следовательно, должно быть полным, и проводить его надо в условиях, исключающих потерю серы в виде летучих соединений. Для этого требуется обработка пробы сильными окислителями, и обязательно в щелочной среде. Если содержание, органических веществ в пробе невелико, то во многих случаях достаточно кипячения пробы после добавления к ней бромной воды. При высоком содержании органических веществ и в присутствии свободной серы и трудноокисляемых веществ обработку надо проводить бромом в присутствии хлороформа или тетрахлорида углерода (в которых растворяется как бром, так и большое число органических веществ и сера) и завершать окисление спеканием со смесью MgO + Na2 CО3 сухого остатка после выпаривания.
Если заранее неизвестно, будут ли органические вещества анализируемой сточной воды полностью окисляться первым способом, рекомендуется провести одно определение обоими способами и при совпадении полученных результатов в дальнейшем, при анализе вод того же типа, применять более простой способ окисления.
Поскольку даже чистые для анализа реактивы часто содержат небольшие количества сульфатов, необходимо проведение холостого опыта и введение поправки на реактивы.
Реактивы
Едкий натр, кристаллический чда и 50%-ный раствор. Растворяют 50 в NaOH чда в дистиллированной воде и разбавляют до 100 мл. Бром чда, насыщенный водный раствор. Соляная кислота чда, концентрированная. Азотная кислота чда, концентрированная. Хлороформ чДа или тетрахлорид углерода чда.
Смесь для окисления. Тщательно перемешивают, растирая в ступке, смесь 2 частей MgO и 1 части Na2 С03 .
Ход определения. Сточная вода содержит только легкоокисляемые в органические вещества. Пробу сразу после отбора консервируют добавлением 3—4 г/л едкого натра.
В коническую колбу вместимостью 300—500 мл наливают в зависимости от предполагаемого содержания серы от 10 до 250 мл тщательно перемешанной пробы, содержащей более 1,5 мг серы. Если пробу не консервировали, к ней прибавляют 1 мл раствора едкого натра. Смесь подогревают на водяной бане и по каплям прибавляют раствор брома до появления неисчезающей жёлтой окраски. Затем нагревают 30 мин, прибавляя раствор брома, если желтая окраска при нагревании исчезает. В случае образования большого количества гидроксида железа (III) смесь фильтруют через стеклянный фильтрующий тигель и осадок тщательно промывают дистиллированной водой.
Горячий раствор нейтрализуют соляной кислотой и после нейтрализации подкисляют 1 мл той же кислоты. Подкисленный раствор нагревают до полного удаления избытка брома и содержимое колбы фильтруют через бумажный беззольный фильтр в стакан вместимостью около 400 мл. Фильтр тщательно промывают дистиллированной водой, фильтрат разбавляют дистиллированной водой до 200 мл и в полученном растворе определяют сульфат-ионы.
Сточная вода содержит трудноокисляемые органические вещества. В коническую колбу вместимостью 300—500 мл наливают, в зависимости от предполагаемого содержания серы, от 10 до 250 мл тщательно перемешанной пробы, содержащей более 1,5 мг серы, прибавляют 1—2 мл раствора едкого натра, 4 мл брома и 5 мл хлороформа или тетрахлорида углерода. Горло колбы закрывают воронкой, перемешанную смесь оставляют при комнатной температуре на 20 мин, вводят 10 мл азотной кислоты и нагревают на водяной бане до удаления избытка брома. Смесь переносят по частям в платиновую чашку, выпаривают на водяной бане досуха и обрабатывают остаток Б мл соляной кислоты, растирая его стеклянной палочкой. Раствор снова выпаривают досуха, прибавляют 0,5—0,7 г смеси карбоната натрия и оксида магния. Смесь тщательно перемешивают палочкой и постеннно нагревают, повышая температуру до тех пор, пока не образуется спекшаяся масса или расплав. Поддерживают эту температуру 20 мин. После охлаждения спекшуюся массу обрабатывают горячей водой и фильтруют в фарфоровую чашку. Остаток в платиновой чашке несколько раз кипятят с дистиллированной водой и фильтруют в ту же чашку, а фильтр тщательно промывают дистиллированной водой. Фильтрат в фарфоровой чашке нейтрализуют соляной кислотой и добавляют 1 мл ее избытка. Содержимое чашки выпаривают на водяной бане досуха. Остаток обрабатывают горячей дистиллированной водой с 1 мл соляной кислоты и отфильтровывают кремневую кислоту; фильтрат собирают в стакан вместимостью примерно 400 мл. После тщательного промывания фильтра горячей дистиллированной водой объем фильтрата доводят приблизительно до 200 мл и в полученном растворе определяют сульфат-ионы.